
Журнал "Гастроэнтерология" Том 56, №1, 2022
Вернуться к номеру
Роль кишкової проникності в прогресуванні неалкогольної жирової хвороби печінки у дітей з ожирінням
Авторы: Yu.M. Stepanov, N.Yu. Zavhorodnia, N.O. Zhyhyr
SI ”Institute Gastroenterology of the National Academy of Medical Sciences of Ukraine“, Dnipro, Ukraine
Рубрики: Гастроэнтерология
Разделы: Клинические исследования
Версия для печати
Численні дослідження останніх років підтвердили підвищення кишкової проникності при розвитку неалкогольної жирової хвороби печінки (НАЖХП) і прогресуванні до неалкогольного стеатогепатиту (НАСГ) і фіброзу печінки. У статті розглянуто поширеність, перебіг та діагностичні критерії педіатричної НАЖХП. Продемонстровано роль підвищеної проникності кишечника в патогенезі НАЖХП. Приділено увагу структурі кишкового бар’єра та можливим методам дослідження його проникності. Проведено огляд сучасних досліджень проникності кишечника при НАЖХП у дорослих і дітей, що підтверджують її ключову роль у прогресуванні НАЖХП. Пошук літератури був проведений в електронних базах даних Scopus, MedLine, EMBASE, Pubmed, Google Scholar тощо.
Many studies in recent years have revealed increased intestinal permeability in the non-alcoholic fatty liver disease (NAFLD) development and progression to nonalcoholic steatohepatitis (NASH) and liver fibrosis. The prevalence, course, and diagnostic criteria of pediatric NAFLD were considered in the article. The role of increased intestinal permeability in the pathogenesis of NAFLD has been demonstrated. Attention was paid to the structure of the intestinal barrier and possible methods for its permeability examination. Current studies of intestinal permeability in NAFLD in adults and children, which confirm its key role in the progression of NAFLD, were reviewed. A literature search was conducted in electronic databases Scopus, MedLine, EMBASE, Pubmed, Google Scholar, etc.
кишкова проникність; неалкогольна жирова хвороба печінки; ожиріння; діти; огляд
intestinal permeability; non-alcoholic fatty liver disease; obesity; children; review
Introduction
Terminology
Epidemiology of NAFLD
The course of the disease
Diagnosis
Pathogenesis of NAFLD
/27.jpg)
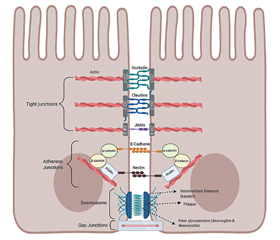
Intestinal barrier structure
/28.jpg)
/29.jpg)
Research overview
Methods for intestinal permeability examination
Conclusions
- Rehm J.L., Connor E.L., Wolfgram P.M., Eickhoff J.C., Reeder S.B., Allen D.B. Predicting hepatic steatosis in a racially and ethnically diverse cohort of adolescent girls. J. Pediatr. 2014. 165(2). 319-325.e1. doi: 10.1016/j.jpeds.2014.04.019.
- Anderson E.L., Howe L.D., Jones H.E., Higgins J.P., Lawlor D.A., Fraser A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS One. 2015. 10(10). e0140908. doi: 10.1371/journal.pone.0140908.
- Yu E.L., Golshan S., Harlow K.E., et al. Prevalence of Nonalcoholic Fatty Liver Disease in Children with Obesity. J. Pediatr. 2019. 207. 64-70. doi: 10.1016/j.jpeds.2018.11.021.
- Tricò D., Caprio S., Rosaria Umano G., et al. Metabolic Features of Nonalcoholic Fatty Liver (NAFL) in Obese Adolescents: Findings From a Multiethnic Cohort. Hepatology. 2018 Oct. 68(4). 1376-1390. doi: 10.1002/hep.30035.
- Younossi Z., Anstee Q.M., Marietti M., et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018. 15(1). 11-20. doi: 10.1038/nrgastro.2017.109.
- Mandala A., Janssen R.C., Palle S., Short K.R., Friedman J.E. Pediatric Non-Alcoholic Fatty Liver Disease: Nutritional Origins and Potential Molecular Mechanisms. Nutrients. 2020. 12(10). 3166. doi: 10.3390/nu12103166.
- Vos M.B., Abrams S.H., Barlow S.E., et al. NASPGHAN Clinical Practice Guideline for the Diagnosis and Treatment of Nonalcoholic Fatty Liver Disease in Children: Recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). J. Pediatr. Gastroenterol. Nutr. 2017. 64(2). 319-334. doi: 10.1097/MPG.0000000000001482.
- D’Adamo E., Castorani V., Nobili V. The Liver in Children With Metabolic Syndrome. Front Endocrinol. (Lausanne). 2019. 10. 514. Published 2019 Aug 2. doi: 10.3389/fendo.2019.00514.
- Jennison E., Byrne C.D. The role of the gut microbiome and diet in the pathogenesis of non-alcoholic fatty liver disease. Clin. Mol. Hepatol. 2021. 27(1). 22-43. doi: 10.3350/cmh.2020.0129.
- Schwimmer J.B., Zepeda A., Newton K.P., et al. Longitudinal assessment of high blood pressure in children with nonalcoholic fatty liver disease. PLoS One. 2014. 9(11). e112569. Published 2014. Nov 24. doi: 10.1371/journal.pone.0112569.
- Holterman A.X., Guzman G., Fantuzzi G., et al. Nonalcoholic fatty liver disease in severely obese adolescent and adult patients. Obesity (Silver Spring). 2013 Mar. 21(3). 591-7. doi: 10.1002/oby.20174. PMID: 23592668.
- Nier A., Brandt A., Conzelmann I.B., Özel Y., Bergheim I. Non-Alcoholic Fatty Liver Disease in Overweight Children: Role of Fructose Intake and Dietary Pattern. Nutrients. 2018. 10(9). 1329. Published 2018 Sep 19. doi: 10.3390/nu10091329.
- Pendyala S., Walker J.M., Holt P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology. 2012. 142(5). 1100-1101.e2. doi: 10.1053/j.gastro.2012.01.034.
- Netto Candido T.L., Bressan J., Alfenas R.C.G. Dysbiosis and metabolic endotoxemia induced by high-fat diet. Nutr. Hosp. 2018 Dec 3. 35(6). 1432-1440. English. doi: 10.20960/nh.1792.
- Plaza-Díaz J., Solís-Urra P., Rodríguez-Rodríguez F., et al. The Gut Barrier, Intestinal Microbiota, and Liver Disease: Molecular Mechanisms and Strategies to Manage. Int. J. Mol. Sci. 2020. 21(21). 8351. Published 2020 Nov 7. doi: 10.3390/ijms21218351.
- Di Ciaula A., Baj J., Garruti G., et al. Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. J. Clin. Med. 2020. 9(8). 2648. Published 2020 Aug 14. doi: 10.3390/jcm9082648.
- Nighot M., Al-Sadi R., Guo S., et al. Lipopolysaccharide-Induced Increase in Intestinal Epithelial Tight Permeability Is Mediated by Toll-Like Receptor 4/Myeloid Differentiation Primary Response 88 (MyD88) Activation of Myosin Light Chain Kinase Expression. Am. J. Pathol. 2017 Dec. 187(12). 2698-2710. doi: 10.1016/j.ajpath.2017.08.005.
- Guo S., Nighot M., Al-Sadi R., Alhmoud T., Nighot P., Ma T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015. 195(10). 4999-5010. doi: 10.4049/jimmunol.1402598.
- Sharifnia T., Antoun J., Verriere T.G., et al. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015. 309(4). G270-G278. doi: 10.1152/ajpgi.00304.2014.
- Liu J., Zhuang Z.J., Bian D.X., et al. Toll-like receptor-4 signalling in the progression of non-alcoholic fatty liver disease induced by high-fat and high-fructose diet in mice. Clin. Exp. Pharmacol. Physiol. 2014 Jul. 41(7). 482-8. doi: 10.1111/1440-1681.12241.
- Mouzaki M., Loomba R. Insights into the evolving role of the gut microbiome in nonalcoholic fatty liver disease: rationale and prospects for therapeutic intervention. Therap. Adv. Gastroenterol. 2019. 12. 1756284819858470. Published 2019 Jun 23. doi: 10.1177/1756284819858470.
- Kisseleva T., Brenner D.A. Hepatic stellate cells and the reversal of fibrosis. J. Gastroenterol. Hepatol. 2006 Oct. 21. Suppl. 3. S84-7. doi: 10.1111/j.1440-1746.2006.04584.x.
- Giorgio V., Miele L., Principessa L., et al. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig. Liver Dis. 2014 Jun. 46(6). 556-60. doi: 10.1016/j.dld.2014.02.010.
- Imajo K., Fujita K., Yoneda M., et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012 Jul 3. 16(1). 44-54. doi: 10.1016/j.cmet.2012.05.012.
- Mouries J., Brescia P., Silvestri A., et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019. 71(6). 1216-1228. doi: 10.1016/j.jhep.2019.08.005.
- Zhou J., Tripathi M., Sinha R.A., Singh B.K., Yen P.M. Gut microbiota and their metabolites in the progression of non-alcoholic fatty liver disease. Hepatoma Res. 2021 Jan 13. 7. 11. doi: 10.20517/2394-5079.2020.134.
- Kirpich I.A., Marsano L.S., McClain C.J. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015. 48(13-14). 923-930. doi: 10.1016/j.clinbiochem.2015.06.023.
- Poeta M., Pierri L., Vajro P. Gut-Liver Axis Derangement in Non-Alcoholic Fatty Liver Disease. Children (Basel). 2017. 4(8). 66. Published 2017 Aug 2. doi: 10.3390/children4080066.
- Mohammad S., Thiemermann C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front Immunol. 2021. 11. 594150. Published 2021 Jan 11. doi: 10.3389/fimmu.2020.594150.
- Chen D., Le T.H., Shahidipour H., Read S.A., Ahlenstiel G. The Role of Gut-Derived Microbial Antigens on Liver Fibrosis Initiation and Progression. Cells. 2019. 8(11). 1324. Published 2019 Oct 27. doi: 10.3390/cells8111324.
- Ilan Y. Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol. 2012. 18(21). 2609-2618. doi: 10.3748/wjg.v18.i21.2609.
- Inzaugarat M.E., Wree A., Feldstein A.E. Hepatocyte mitochondrial DNA released in microparticles and toll-like receptor 9 activation: A link between lipotoxicity and inflammation during nonalcoholic steatohepatitis. Hepatology. 2016. 64(2). 669-671. doi: 10.1002/hep.28666.
- Alisi A., Manco M., Devito R., Piemonte F., Nobili V. Endotoxin and plasminogen activator inhibitor-1 serum levels associated with nonalcoholic steatohepatitis in children. J. Pediatr. Gastroenterol. Nutr. 2010 Jun. 50(6). 645-9. doi: 10.1097/MPG.0b013e3181c7bdf1.
- Wong V.W., Wong G.L., Chan H.Y., et al. Bacterial endotoxin and non-alcoholic fatty liver disease in the general population: a prospective cohort study. Aliment. Pharmacol. Ther. 2015. 42(6). 731-40. doi: 10.1111/apt.13327.
- Miele L., Valenza V., La Torre G., et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009. 49(6). 1877-87. doi: 10.1002/hep.22848.
- Luther J., Garber J.J., Khalili H., et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol. Gastroenterol. Hepatol. 2015 Mar. 1(2). 222-232. doi: 10.1016/j.jcmgh.2015.01.001.
- Schoultz I., Keita Å.V. The Intestinal Barrier and Current Techniques for the Assessment of Gut Permeability. Cells. 2020. 9(8). 1909. Published 2020 Aug 17. doi: 10.3390/cells9081909.
- Albillos A., de Gottardi A., Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020 Mar. 72(3). 558-577. doi: 10.1016/j.jhep.2019.10.003.
- Dai X., Hou H., Zhang W., et al. Microbial Metabolites: Critical Regulators in NAFLD. Front Microbiol. 2020. 11. 567654. Published 2020 Oct 7. doi: 10.3389/fmicb.2020.567654.
- Chopyk D.M., Grakoui A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology. 2020. 159(3). 849-863. doi: 10.1053/j.gastro.2020.04.077.
- Van Itallie C.M., Anderson J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014. 36. 157-65. doi: 10.1016/j.semcdb.2014.08.011.
- Guo S., Al-Sadi R., Said H.M., Ma T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 2013. 182(2). 375-387. doi: 10.1016/j.ajpath.2012.10.014.
- Plaza-Díaz J., Solis-Urra P., Aragón-Vela J., Rodríguez-Rodríguez F., Olivares-Arancibia J., Álvarez-Mercado A.I. Insights into the Impact of Microbiota in the Treatment of NAFLD/NASH and Its Potential as a Biomarker for Prognosis and Diagnosis. Biomedicines. 2021. 9(2). 145. Published 2021 Feb 3. doi: 10.3390/biomedicines9020145.
- Nier A., Engstler A.J., Maier I.B., Bergheim I. Markers of intestinal permeability are already altered in early stages of non-alcoholic fatty liver disease: Studies in children. PLoS One. 2017. 12(9). e0183282. Published 2017 Sep 7. doi: 10.1371/journal.pone.0183282.
- De Munck T.J.I., Xu P., Verwijs H.J.A., et al. Intestinal permeability in human nonalcoholic fatty liver disease: A systematic review and meta-analysis. Liver Int. 2020. 40(12). 2906-2916. doi: 10.1111/liv.14696.
- Nobili V., Alisi A., Cutrera R., et al. Altered gut-liver axis and hepatic adiponectin expression in OSAS: novel mediators of liver injury in paediatric non-alcoholic fatty liver. Thorax. 2015 Aug. 70(8). 769-81. doi: 10.1136/thoraxjnl-2015-206782.
- Hendy O.M., Elsabaawy M.M., Aref M.M., Khalaf F.M., Oda A.M.A., El Shazly H.M. Evaluation of circulating zonulin as a potential marker in the pathogenesis of nonalcoholic fatty liver disease. APMIS. 2017 Jul. 125(7). 607-613. doi: 10.1111/apm.12696.
- Chwist A., Hartleb M., Lekstan A., Kukla M., Gutkowski K., Kajor M. A composite model including visfatin, tissue polypeptide-specific antigen, hyaluronic acid, and hematological variables for the diagnosis of moderate-to-severe fibrosis in nonalcoholic fatty liver disease: a preliminary study. Pol. Arch. Med. Wewn. 2014. 124(12). 704-12. doi: 10.20452/pamw.2558.
- Volynets V., Küper M.A., Strahl S., et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012 Jul. 57(7). 1932-41. doi: 10.1007/s10620-012-2112-9.
- Troisi J., Pierri L., Landolfi A., et al. Urinary Metabolomics in Pediatric Obesity and NAFLD Identifies Metabolic Pathways/Metabolites Related to Dietary Habits and Gut-Liver Axis Perturbations. Nutrients. 2017. 9(5). 485. Published 2017 May 11. doi: 10.3390/nu9050485.
- Guercio Nuzio S., Di Stasi M., Pierri L., et al. Multiple gut-liver axis abnormalities in children with obesity with and without hepatic involvement. Pediatr. Obes. 2017 Dec. 12(6). 446-452. doi: 10.1111/ijpo.12164.
- Pacifico L., Bonci E., Marandola L., Romaggioli S., Bascetta S., Chiesa C. Increased circulating zonulin in children with biopsy-proven nonalcoholic fatty liver disease. World J. Gastroenterol. 2014. 20(45). 17107-17114. doi: 10.3748/wjg.v20.i45.17107.
- Jin R., Willment A., Patel S.S., et al. Fructose induced endotoxemia in pediatric nonalcoholic Fatty liver disease. Int. J. Hepatol. 2014. 2014. 560620. doi: 10.1155/2014/560620.
- Çakır M., Aksel İşbilen A., Eyüpoğlu İ., et al. Effects of long-term synbiotic supplementation in addition to lifestyle changes in children with obesity-related non-alcoholic fatty liver disease. Turk. J. Gastroenterol. 2017 Sep. 28(5). 377-383. doi: 10.5152/tjg.2017.17084.
- Loffredo L., Zicari A.M., Perri L., et al. Does Nox2 Overactivate in Children with Nonalcoholic Fatty Liver Disease? Antioxid. Redox Signal. 2019 Apr 1. 30(10). 1325-1330. doi: 10.1089/ars.2018.7596.
- Rosso C., Caviglia G.P., Younes R., et al. Circulating Zonulin is Related to Hepatic Necroinflammation in Patients with Non Alcoholic Fatty Liver Disease. Clin. Lab. 2020 Apr 1. 66(4). doi: 10.7754/Clin.Lab.2019.190922.
- Guercio Nuzio S., Di Stasi M., Pierri L., et al. Multiple gut-liver axis abnormalities in children with obesity with and without hepatic involvement. Pediatr. Obes. 2017 Dec. 12(6). 446-452. doi: 10.1111/ijpo.12164.
- Zhou X., Han D., Xu R., et al. A model of metabolic syndrome and related diseases with intestinal endotoxemia in rats fed a high fat and high sucrose diet. PLoS One. 2014. 9(12). e115148. Published 2014 Dec 11. doi: 10.1371/journal.pone.0115148.
- Grootjans J., Thuijls G., Verdam F., Derikx J.P., Lenaerts K., Buurman W.A. Non-invasive assessment of barrier integrity and function of the human gut. World J. Gastrointest. Surg. 2010. 2(3). 61-69. doi: 10.4240/wjgs.v2.i3.61.
- Kolodziejczyk A.A., Zheng D., Shibolet O., Elinav E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019. 11(2). e9302. doi: 10.15252/emmm.201809302.