
Журнал "Гастроэнтерология" Том 53, №2, 2019
Вернуться к номеру
Погляд на фіброз печінки у світлі розуміння сучасних механізмів його розвитку
Авторы: Колеснікова О.В.
ДУ «Національний інститут терапії ім. Л.Т. Малої НАМН України», м. Харків, Україна
Рубрики: Гастроэнтерология
Разделы: Справочник специалиста
Версия для печати
У статті подані сучасні погляди на механізми формування фіброзу печінки. Особливу увагу приділено ролі й функції зірчастих клітин печінки, процесам регенерації пошкодженої печінки. Висвітлений зв’язок розвитку фіброзу печінки з генетичними й негенетичними факторами. На прикладі розвитку фіброзу печінки в пацієнтів із неалкогольною жировою хворобою печінки показано в динаміці неінвазивного моніторингу позитивний вплив препарату Схізандрин® на зменшення ступеня фіброзних змін у печінці в цієї категорії пацієнтів. Останнє відзначається на тлі нормалізації метаболічних показників у пацієнтів із неалкогольною жировою хворобою печінки протягом 6 місяців терапії.
В статье представлены современные взгляды на механизмы формирования фиброза печени. Особое внимание уделено роли и функции звездчатых клеток печени, процессам регенерации поврежденной печени. Освещена связь развития фиброза печени с генетическими и негенетическими факторами. На примере развития фиброза печени у пациентов с неалкогольной жировой болезнью печени показано в динамике неинвазивного мониторинга положительное влияние препарата Схизандрин® на уменьшение степени фиброзных изменений в печени у этой категории пациентов. Последнее отмечается на фоне нормализации метаболических показателей у пациентов с неалкогольной жировой болезнью печени в течение 6 месяцев терапии.
The article presents modern views on the mechanisms of formation of liver fibrosis. Particular attention is paid to the role and function of hepatic stellate cells, the processes of regeneration of damaged liver. The correlation between the development of liver fibrosis and genetic and non-genetic factors is described. The positive effect of Schisandrin® on reducing the degree of fibrotic changes in the liver in this category of patients is shown in the dynamics of non-invasive monitoring on the example of liver fibrosis development in non-alcoholic fatty liver disease. The latter is noted against the background of the normalization of metabolic parameters in patients with non-alcoholic fatty liver disease during 6 months of therapy.
фіброз печінки; механізми розвитку; неалкогольна жирова хвороба печінки; маркери фіброзу печінки; Схізандрин®
фиброз печени; механизмы развития; неалкогольная жировая болезнь печени; маркеры фиброза печени; Схизандрин®
liver fibrosis; mechanisms of development; non-alcoholic fatty liver disease; markers of liver fibrosis; Schisandrin®
Вступ
Понад 2000 років тому Еразістрат (Erasistratos), прихильник Олександрійської медичної школи, висловив припущення про зернисту індурацію печінки як про причину водянки (асциту). Із того часу цікавість медиків до патофізіології ущільнення печінки все більше зростала. У 1819 році Р. Лаеннек (R.T. Laёnnec) описав цей стан як окреме захворювання і назвав його «цироз». У подальшому Р. Карсвелем (R. Carswell) у 1838 році було показано, що фіброз є характерною рисою цирозу печінки (ЦП). Через п’ять років, у 1843 р., Й. Мюллер (J. Müller) зазначив, що хронічне запалення призводить до гіпертрофії міжчасточкової сполучної тканини. У 1858 році Р. Вірхов (R. Virchow) виявив, що джерелом збільшення кількості сполучної тканини є клітини, а в 1872 році В. Легг (W. Legg) показав, що некроз печінкових клітин, будучи невід’ємною частиною репаративного процесу, першим запускає процес відкладення екстрацелюлярного матриксу (ЕЦМ) [1]. Із 50-х років ХХ століття робляться спроби пояснити механізми розвитку фіброзу печінки (ФП). Так, у 1954 році В. Хартрофт (W.S. Hartroft) пропонує теорію розвитку ФП, засновану на руйнуванні некротизованої паренхіми з подальшим ущільненням строми в септи [2]. І тільки в 1978 році група патологів, очолювана P.P. Anthony і K.G. Ishak, уперше вводить визначення й номенклатуру ФП. Фіброз описується як «наявність надмірної кількості колагену у зв’язку з новоутворенням волокон» [3]. Фіброз став обов’язковим критерієм цирозу печінки, що розглядався як «перетворення нормальної архітектоніки печінки в структурно-аномальні вузли». Незважаючи на спрощене визначення фіброзу, що не включало важливі неколагенові компоненти ЕЦМ, воно вказувало саме на біосинтез ЕЦМ як основний патогенетичний шлях, що веде до колапсу паренхіми печінки. Ці факти, безсумнівно, стимулювали подальше вивчення патогенезу ФП і ідентифікацію клітинних типів, залучених до процесу продукції ЕЦМ. Одними з перших у своїх роботах Мак-Гі (J.O. McGee), Патрік (R.S. Patrick), Кент (G. Kent) і співавт. вказали на участь у синтезі колагену перисинусоїдальних ліпоцитів, що містять вітамін А. Уперше цей тип клітин був виявлений вченим Ф. Боллом (F. Boll) у 1869 р. [4] і потім був детально описаний С. Купфером (C.W. Kupffer) у 1876 р. [5]. Відкриття у 1952 р. нового виду клітин, названих жирозберігальними, пов’язане з ім’ям Т. Іто (T. Ito) [6]. Майже через 20 років, у 1971 р., К. Вейк (K. Wake) наведе докази того, що вони ідентичні зірчастим клітинам, відкритим Купфером [7].
Протягом багатьох років перицитам, що розташовані в субендотеліальному просторі Діссе й оточують шар синусоїдальних ендотеліоцитів, давали різні назви: жирозберігальні клітини, клітини, що містять вітамін А, ліпоцити, парасинусоїдальні клітини, клітини Іто, арахноцити тощо.
У 1996 році групою з 98 дослідників у цій галузі був запропонований термін hepatic stellate cells (зірчасті клітини печінки (ЗКП)), який був прийнятий науковим товариством [8].
Вивчення структури, функцій, патофізіології ЗКП стало можливим завдяки D. Knook і його групі дослідників, які в 1982 році запропонували методи ізоляції й культивування цих клітин із печінки щурів. Вони об’єднали техніку послідовної проназно-колагеназної перфузії печінки з цетрифугуванням суспензії клітин у градієнті щільності з метою накопичення ЗКП у верхньому шарі градієнта метризаміду. Цей метод був успішно адаптований для виділення ЗКП із ділянок нормальної людської печінки [9]. З того часу стало можливим проведення численних досліджень, які показали ключову роль ЗКП у патогенезі ФП, синтезі колагену, глікозаміногліканів, гіалуронату, фібронектину, тенасцину. Відкриття морфологічно схожих клітин в інших органах, наприклад у нирках, підшлунковій залозі, кишечнику, привело до розвитку концепції про дифузійну систему стелатних клітин у ссавців.
Метою цієї публікації є подання існуючих поглядів на механізми розвитку фіброзу печінки на підставі сучасних знань, а також можливість продемонструвати «управління» процесом фіброутворення за допомогою моніторингу та впливу на патогенетичні механізми його формування.
Що ми знаємо сьогодні про фіброз печінки?
Сучасний стан питання
Експериментальні дослідження останніх 20 років значно розширили й деталізували знання про структуру й закономірності відкладення екстрацелюлярного матриксу в нормальній і фіброзній тканині печінки; клітинні джерела різних матричних компонентів; стимуляцію синтезу ЕЦМ (фіброгенезу) цитокінами й факторами росту й регуляцію матричної деградації (фіролізу); генетичні фактори, що сприяють розвитку фіброзу; терапевтичні можливості, що ґрунтуються на успішних результатах численних експериментів.
Фіброз печінки є процесом, що характеризується:
— значним (до 10 разів) збільшенням кількості ЕЦМ, що включає декілька типів колагену, структурні глікопротеїни, сульфатовані протеоглікани (глікозаміноглікани) і гіалуронат;
— гістологічною перебудовою з відкладенням матриксу переважно в перивенулярній зоні 3 ацинусів у субендотеліальному просторі Діссе, що веде до формування неповноцінної субендотеліальної базальної мембрани, створюючи додатковий бар’єр між гепатоцитами й печінковими синусоїдами (капіляризація синусоїдів);
— змінами будови ЕЦМ;
— змінами ультраструктури колагенів (наприклад, рівень гідроксилювання проліну й лізину), глікопротеїнів (варіації в структурі вуглеводню), протеогліканів (рівень сульфатування бічних ланцюгів глікозаміногліканів) у поєднанні з варіантами з’єднання молекул ЕЦМ.
Розвиток фіброзу — активний біосинтетичний процес, характерною рисою якого є стимуляція продукції матриксу портальними або перибіліарними фібробластами, а особливо міофібробластами, що розташовуються в субендотеліальному просторі Діссе.
Роль і функції зірчастих клітин печінки
ЗКП (НSC) — це періцити печінки, що мають дендрити. Вони належать до дифузної системи стеллатних клітин організму, становлять близько 1/3 непаренхіматозної клітинної популяції (клітини Купфера, ендотеліальні клітини), 1,4 % об’єму печінки і приблизно 15 % всіх клітин печінки, включаючи гепатоцити. В здоровій печінці щура — індекс зірчастих клітин печінки. Веретеноподібне тіло ЗКП розміром 700 μм3 містить численні вакуолі, багаті на тригліцериди, у яких містяться метаболіти вітаміну А (ретиноїди) у розчиненому вигляді. У ЗКП перебуває близько 85 % вітаміну А печінки. Нещодавно відкриті додаткові функції цих клітин: 1) їх роль як антиген-репрезентативних клітин; 2) їх участь як CD133+ — клітин-попередників у диференціюванні попередників ендотеліальних клітин і гепатоцитів, що відіграють важливу роль у регенерації й репарації печінки; 3) їх залучення в ендоцитоз апоптотичних паренхіматозних клітин; 4) секреція матриксних металопротеїназ (MMP), їх тканинних інгібіторів (TIMP) і факторів росту; 5) підтримання печінкової регенерації за рахунок стимуляції проліферації гепатоцитів за допомогою рецептора нейротропіну р75; 6) регуляція ангіогенезу й судинного ремоделювання шляхом секреції ангіогенних факторів, таких як фактор росту ендотелію судин, ендотелін-1 (ET-1), інсуліноподібний фактор росту II, нейротропіну й еритропоетину; 7) гемодинамічні функції, що полягають у скороченні синусоїдів у відповідь на контракцію активованих ЗКП, що індукуються тромбоксаном, простагландином F2, ангіотензином II, вазопресином, ЕТ-1.
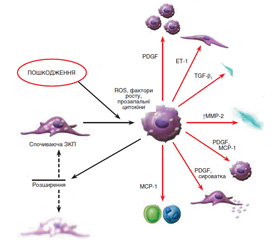
Деякі з цих функцій не відзначені в ЗКП у спокійному стані, але спостерігаються при активації цих клітин. Формування міофібробластів із ЗКП відбувається в декілька послідовних стадій, що ініціюються некрозом печінкових клітин у результаті дії токсичних, імунологічних агентів тощо (рис. 1). У результаті активуються ЗКП, що локалізуються в безпосередній близькості до гепатоцитів.
/69-1.jpg)
Активація ЗКП призводить до експресії α-гладеньком’язового актину, десміну й гелсоліну й паралельного зниження гліального фібрилярного кислотоутворюючого протеїну (GFAP), а також до втрати великої кількості жирових крапель і ретиноїдів, посилення контрактильності, експресії й секреції матриксних компонентів.
Процес активації включає проліферацію й фенотипове трансформування ЗКП у міофібробласти. Цей механізм вважається центральним у розвитку фіброзу. Трансформовані зі стелатних клітин міофібробласти мають здатність не тільки до синтезу матриксу, але і до експресії й секреції багатьох про- і протизапальних цитокінів і факторів росту.
Механізм активації й трансформування ЗКП у міофібробласти можна подати у вигляді моделі триступеневого каскаду, що починається з передзапальної фази шляхом прямої паракринної активації зірчастих клітин некротизованими гепатоцитами з виділенням цитокінів і одночасним зниженням гепарину сульфату на поверхні гепатоцитів, що має властивість інгібування мітозу. У запальну фазу активовані ЗКП, паракринно стимульовані лейкоцитами, тромбоцитами, активованими клітинами Купфера, синусоїдальними ендотеліоцитами, гепатоцитами, трансформуються в міофібробласти. Наступна постзапальна фаза характеризується виділенням фіброгенних цитокінів із міофібробластів і взаємодією матриксних компонентів. Деякі з цих цитокінів стимулюють міофібробласти автокринним шляхом і ЗКП — паракринним. Отже, у постзапальній стадії відбувається підтримка процесу фіброгенезу навіть після закінчення або елімінації перших двох фаз.
Активація й трансформування зірчастих клітин — це результат вираженої взаємодії резидентних клітин печінки з нерезидентними (рис. 1). Більшість медіаторів є вільними радикалами, що продукуються клітинами Купфера і лейкоцитами. Серед пептидних медіаторів основним профіброгенним цитокіном є трансформуючий ростовий фактор бета (TGF-β). Крім того, у процес фіброгенезу також залучені тромбоцитарні фактори росту (PDGF) — PDGF-B і PDGF-D, ET-1, декілька факторів росту фібробластів, фактор некрозу пухлини (ФНП-α), адипоцитокіни (лептин, адипонектин) та інші.
TGF-β секретується у високомолекулярній латентній формі зірчастими клітинами (міофібробластами), синусоїдальними ендотеліоцитами, клітинами Купфера й виділяється тромбоцитами й гепатоцитами. Він не тільки ініціює перетворення ЗКП у міофібробласти, але й посилює експресію матриксних генів, знижуючи вироблення матриксних металопротеаз, і збільшує вироблення їх тканинних інгібіторів, а також індукує апоптоз гепатоцитів та інгібує проліферацію печінкових клітин.
Екстрацелюлярна активація латентного TGF-β протеазами, вільними радикалами, тромбоспондином типу 1, інтегринами αβ1 і αβ8 є важливим етапом у регуляції біодоступності TGF-β. Застосування антагоністів TGF-β або інгібування каскаду реакцій, що ними запускається, значно гальмує й навіть інгібує активацію ЗКП і дає тривалий антифібротичний ефект. Відзначено, що відповідь TGF-β змінюється в процесі перетворення ЗКП у міофібробласти, досягаючи часткової нечутливості до міофібробластів. Це спостереження дає підставу припустити, що TGF-β відіграє важливу роль в ініціації активації ЗКП in vivo, але менше впливає на процес трансформування.
Справжні дослідження приділяють велику увагу контролю активації ЗКП на рівні транскрипції. Велика кількість транскрипційних медіаторів і епігенетичних механізмів (ацетилювання гістонів, метилювання білків-промоутерів) залучені в процес активації [13].
У зв’язку з морфологічною й функціональною інтралобулярною (зональною) гетерогенністю ЗКП процес активації й трансформування (трансдиференціації) in situ різниться топографічно. Це залежить також і від зональної уразливості гепатоцитів. Найбільш чутлива до ураження алкоголем 3-тя зона ацинуса, тому при токсичній дії алкоголю фіброз починається саме з цієї зони.
Гетерогенність ЗКП/міофібробластів пов’язана не тільки з їх локалізацією, але й з різними джерелами. Про це свідчать морфологічні й функціональні відмінності, а також відповідь на фактори росту. Наприклад, ЗКП експресує невральний фактор GFAP, молекули судинної клітинної адгезії 1, білок цитоскелета десмін, що практично відсутні в міофібробластах. З іншого боку, виключно міофібробласти синтезують матриксний протеїн фібулін-2.
Додатковим маркером відмінності ЗКП від інших міофібробластів печінки може бути рилін, білок ЕЦМ, що присутній у спочиваючих і активованих зірчастих клітинах, але не виявлений у міофібробластах. Відзначено, що перибіліарні, паренхіматозні, судинні фіброгенні клітини по-різному експресують трансгени (α-гладеньком’язовий актин і колаген I), що підтверджує їх функціональну гетерогенність.
Отже, питання про зв’язок ЗКП і міофібробластів залишається не до кінця зрозумілим. Передбачається наявність декількох типів міофібробластоподібних клітин. Їх будова й функції можуть вказувати й на інші шляхи формування фіброзних змін (окрім трансдиференції ЗКП) і залежати від природи захворювання.
Роль інших клітин у процесах фіброгенезу печінки
Крім ЗКП, фіброгенний потенціал мають фібробласти, що походять із портальних судин невеликого розміру, які здатні до проліферації навколо проток печінки й накопичення колагену. Доведено, що ЗКП і портальні міофібробласти диференціюються в специфічні клітини-маркери й відповідають на апоптотичні стимули. Цікаво, що порівняльне вивчення культур тканини показало більш швидку проліферацію активованих ЗКП порівняно з портальними фібробластами, можливо, тому вони становлять переважну більшість у популяції міофібробластів при фіброзі печінки.
У цілому ЗКП є основними клітинами, що беруть участь в фіброгенезі в перицентральній ділянці, а портальні міофібробласти домінують у випадку пошкодження навколо портальних трактів.
В останніх дослідженнях на експериментальних тваринах і на людях було показано, що частина міофібробластів печінки може походити зі стовбурових клітин кісткового мозку [14]. Культури CD34, CD38 гемопоетичних стовбурових клітин із різними факторами росту обумовлюють проліферацію ЗКП і міофібробластів кісткового мозку. Останні здатні інфільтрувати печінку, обумовлюючи ремодулювання її тканини. Відзначено також, що ці клітини широко розподілені по ходу сполучної тканини при вираженому фіброзі. Саме ці дані підтверджують, що клітини, які походять із кісткового мозку, можуть бути джерелом фіброгенних клітин у пошкодженій печінці. Більше того, результати одного з досліджень на мишах показали, що пересадка генетично модифікованого кісткового мозку від реципієнта з клінікою фіброзу печінки відбилася на генотипі миші-донора. Це дослідження дає підстави вважати, що залучені з кісткового мозку клітини впливають на фіброз шляхом експресії, синтезу й секреції колагену I. Це дослідження не показало точні механізми переходу клітин кісткового мозку в печінку; також поки відсутні дані про перетворення цих клітин безпосередньо в міофібробласти або через стадію ЗКП. Однак є припущення, що вони можуть походити з CD45+ фіброцитів у присутності TGF-β1 [15].
Особлива увага в даний час приділяється епітеліально-мезенхімальній трансформації епітеліальних клітин жовчних проток і гепатоцитів як можливого джерела субпопуляції міофібробластів при ФП. Ці додаткові відомості розширюють механізми патогенезу ФП за рахунок підвищення рівня знань про кількість матрикс-синтезуючих міофібробластів в ураженій печінці.
Регенерація пошкодженої печінки й формування фіброзу печінки
Якщо раніше вважалося, що ФП і кінцева його стадія, цироз, — це пасивний і незворотний процес, який являє собою деградацію печінкової паренхіми й заміщення її тканиною, багатою колагеном, то в даний час запропонована модель, у якій заміщення сполучною тканиною розглядається як репаративний процес — відповідь на хронічне ураження печінки.
Репарація пошкоджених тканин є фундаментальним біологічним процесом, що полягає в упорядкованому заміщенні загиблих або пошкоджених клітин, механізмом, необхідним для життєдіяльності. Дефект тканин може бути результатом дії різних гострих або хронічних стимулів, таких як інфекційні або токсичні агенти, автоімунні реакції або механічний вплив.
Репаративний процес зазвичай має дві різні стадії: а) фаза регенерації, при якій пошкоджені клітини заміщаються клітинами такого ж типу, не залишаючи слідів патологічного процесу; б) так звана фаза фіброплазії, або фіброзу, при якій паренхіматозна тканина заміщується сполучною. Будучи від початку позитивним, процес загоєння стає патологічним в умовах значного, безконтрольного ремоделювання екстрацелюлярного матриксу й формування рубцевої тканини. У деяких випадках це може призвести до недостатності органа й смерті.
На відміну від гострих запальних процесів, що характеризуються швидкою реакцією з боку судин, набряком, нейтрофільною інфільтрацією, патологічний фіброз зазвичай є результатом хронічних запальних реакцій (протягом тижнів або місяців) і являє собою процес, у якому запалення, тканинна деструкція й процеси регенерації відбуваються одночасно. Незалежно від етіологічних факторів і клінічних відмінностей, для фіброзних порушень будь-якої локалізації характерна наявність персистуючого агента, що підтримує продукцію факторів росту, протеолітичних ферментів, ангіогенних факторів і фіброгенних цитокінів, які стимулюють відкладення сполучнотканинних елементів, прогресування ремоделювання й руйнування нормальної архітектоніки тканини.
У нормі у відповідь на пошкодження тканини уражені епітеліальні/ендотеліальні клітини виробляють медіатори запалення, що ініціюють каскад реакцій коагуляції-антифібринолізу, який запускає механізм тромбоутворення й тимчасової активації ЕЦМ. Під впливом компонентів ЕЦМ відбуваються агрегація тромбоцитів, тромбоутворення, гемостаз. Далі дегрануляція тромбоцитів призводить до вазодилатації й підвищення проникності судин, у той час як стимульовані міофібробласти (колаген-секретуючі α-SMA+ фібробласти) і епітеліальні/ендотеліальні клітини продукують матриксні металопротеїнази, що порушують цілісність базальної мембрани й привертають достатню кількість клітин запалення до місця пошкодження. Епітеліальні/ендотеліальні клітини також секретують фактори росту, цитокіни, хемокіни, що стимулюють проліферацію й залучення нейтрофілів, макрофагів, Т- і В-клітин, еозинофілів на ранніх етапах загоєння. Під час цієї фази первинної міграції лейкоцитів активовані макрофаги й нейтрофіли елімінують залишки тканини, мертві клітини й будь-які організми, що завдають шкоди. Вони також підсилюють процес репарації за рахунок продукції цитокінів і хемокінів, що є мітогенними й хемотаксичними для ендотеліальних клітин. Ендотеліоцити обмежують осередок ураження й формують нові кровоносні судини шляхом міграції до центру вогнища. Згодом активуються Т-клітини, що секретують профіброгенні цитокіни, такі як інтерлейкін-13 і TGF-β, а також макрофаги й фібробласти.
Активовані фібробласти під час міграції вздовж мережі фібрину в осередок пошкодження трансформуються в α-SMA-секретуючі міофібробласти. Міофібробласти, як згадувалося раніше, можуть утворюватися з місцевих мезенхімальних клітин, активуватися з кісткового мозку (де вони відомі як фіброцити) або трансформуватися з епітеліальних клітин за рахунок епітеліально-мезенхімальної трансформації.
Далі йде фаза дозрівання й ремоделювання, коли активовані міофібробласти підсилюють контракцію осередку ураження, колагенові волокна стають більш організованими, відновлюються кровоносні судини, рубцева тканина елімінується, а епітеліальні/ендотеліальні клітини діляться й мігрують уздовж базального шару, формуючи епітелій/ендотелій. Так закінчується фізіологічний процес регенерації.
Однак при повторюваному пошкодженні хронічне запалення, некроз тканини і постійний процес репарації призводять до перманентної активності міофібробластів, надмірної акумуляції компонентів ЕЦМ (таких як гіалуронова кислота, фібронектин, протеоглікани, інтерстиціальні колагени), тим самим формується сполучнотканинний рубець.
Кількість колагену, що відкладається фібробластами, безперервно регулюється за рахунок його синтезу й катаболізму. Цей процес контролюється різними матриксними металопротеїназами і їх тканинними інгібіторами, що продукуються гранулоцитами, макрофагами, епідермальними клітинами й міофібробластами. Зрушення між цими двома механізмами (синтез і розпад) регулюють збільшення або зменшення кількості колагену в осередку ураження.
Однак збільшений пул мезенхімальних клітин призводить до дисбалансу, і в фазу ремоделювання продукція колагену починає перевищувати його розпад. Хоча прийнято вважати, що запалення зазвичай передує фіброзу, результати експериментальних досліджень показали, що фіброз не обов’язково йде за запаленням у всіх випадках, і це дозволяє припустити, що механізми регуляції фіброгенезу відрізняються від таких при запаленні. Можливо, цим пояснюється недостатня ефективність протизапальної терапії в лікуванні фіброзних захворювань.
Деякі аспекти молекулярних механізмів фіброзу печінки
Останніми роками знання про клітинні й молекулярні механізми ФП значно розширилися. Кардинальні зміни в розумінні процесів фіброзу базуються на використанні численних експериментальних модельних систем. Наочно показані фази запалення й репарації в печінці, продемонстровано взаємодію між ендотеліальними клітинами, запальними медіаторами, міофібробластами, компонентами ЕЦМ у процесі регенерації печінкової тканини у ссавців.
Сьогодні стало відомо, що перебіг ФП залежить як від генетичних чинників, так і від чинників навколишнього середовища (табл. 1).
/72-1.jpg)
Дослідження, проведені на моделях печінкового фіброзу в мишей, виявили ключові гени, що опосередковують розвиток ФП. До генів, що регулюють гепатоцелюлярний апоптоз і/або некроз, належать Bcl-xL, Fas. Вони впливають на пошкодження печінки й розвиток відповіді у вигляді фіброгенезу. IL-1β, IL-10 і IL-13, IFN-γ, SOCS-1 і остеопонтин віднесені до генів, які не тільки регулюють запальну відповідь на пошкодження, але й визначають фіброгенетичні реакції [16–18].
Показано, що NADPH-оксидаза регулює як запальну активність, так і депозицію молекул екстрацелюлярного матриксу. Серед факторів фіброгенетичного зростання, таких як TGF-β1, фактор росту фібробластів, вазоактивні субстанції (ангіотензин II, норадреналін) і адипокіни (лептин, адипонектин), кожен сам по собі необхідний для розвитку фіброзу в цілому. Нарешті, усунення або зворотний розвиток колагену після припинення шкідливої дії на печінку регулюється тканинними інгібіторами металопротеїназ (TIMP-1) і TGF-β1.
Зіставлення результатів генетичних досліджень із клінічним матеріалом продемонструвало роль генетичного поліморфізму в прогресуванні ФП у пацієнтів із хронічними захворюваннями печінки.
При алкогольних захворюваннях печінки до цих генів можуть бути віднесені ті, які кодують активність ферментів, що метаболізують алкоголь, і сучасних експепротеїнів, залучених в утворення токсичних для печінки субстанцій. Так, поліморфізм генів, що кодують алкогольдегідрогеназу, альдегіддегідрогеназу і цитохром Р450, забезпечує індивідуальну чутливість до розвитку алкоголізму, і їх роль у прогресуванні захворювання печінки все ж таки залишається суперечливою. Варіація генів, що кодують запальні медіатори, такі як ФНП-α, IL-1b, IL-10, цитотоксичний Т-лімфоцитарний антиген 4, ліпополісахаридний рецептор CD-14 і антиоксиданти (такі як супероксиддисмутаза), може впливати на прогресування алкогольного захворювання печінки.
При хронічних холестатичних захворюваннях печінки, таких як первинний біліарний цироз печінки, поліморфізм IL-1b, рецепторів антагоністів IL-1, гена, що кодує ФНП-α, асоціюється з більш швидким прогресуванням хвороби. Деякі алелі гена аполіпротеїну Е впливають на відповідь на терапію первинного біліарного цирозу з використанням урсодезоксихолевої кислоти, і це підтверджує, що генетичний поліморфізм може бути предиктором терапевтичної відповіді [19].
При хронічних вірусних гепатитах С також є генетичні варіації, відповідальні за персистенцію HCV-інфекції, відповідь на антивірусну терапію й прогресування захворювання печінки. Поліморфізм генів, залучених в імунну відповідь на HCV-інфекцію, таких як асоційовані з транспортом і процесією антигену 2, манозозв’язуючий лектин, специфічні HLA-II алелі, агоністи фіброгенезу (ангіотензин і TGF-β1), визначає прогресування фіброзу. Значення для фіброгенезу гетерозиготності за C282Y мутації гена гематохроматозу в пацієнтів із хронічним вірусним гепатитом С є суперечливим. Нарешті, суперечливими залишаються до цього часу фактори, що визначають прогресування неалкогольного стеатогепатиту й поліморфізм медіаторів фіброгенезу, таких як ангіотензин і TGF-β1, які можуть бути асоційовані з більш тяжким перебігом захворювання.
Після дії провокуючого фактора паренхіматозні клітини починають регенерувати й заміщати некротизовані або ті, що зазнали апоптозу, гепатоцити. Цей процес перебігає паралельно із запальною відповіддю й обмеженим накопиченням білків екстрацелюлярного матриксу. У випадку персистенції пошкоджувального фактора регенерація сповільнюється й гепатоцити заміщуються надмірною кількістю білків екстрацелюлярного матриксу, включно з фібрилярним колагеном, розподіл якого залежить від пошкоджувального фактора. Зміни кількісного та якісного складу екстрацелюлярного колагенового матриксу (ЕКМ) характеризують ФП. При виражених стадіях фіброзу печінка містить приблизно до 10 разів більше ЕКМ, ніж у нормі, включаючи колаген (1, 3 і 4-го типів), фібронектин, ундулін, еластин, ламінін, гіалурон і протеоглікани. Зниження деградації компонентів, а також підвищення синтезу елементів ЕКМ призводить до активного накопичення останнього. Зниження швидкості виведення ЕКМ і молекул металопротеїназ в основному є наслідком перевивільнення їх специфічних інгібіторів (TIMPs).
У підсумку стає очевидним, що стадії регенерації й фіброгенезу в печінці включають запалення, формування тимчасового тромбу з подальшою інвазією й проліферацією запальних і матрикс-продукуючих клітин і, нарешті, остаточне відновлення або формування рубця (септи). При повторному (хронічному) пошкодженні відкладення матриксу превалює над його ресорбцією, що викликано дисбалансом між фіброгенезом і фібринолізом і призводить до формування рубця. При цьому важливу роль може відігравати недостатня або повільна регенерація, що сприяє збільшенню вільного простору для відкладення матриксу. При прогресуванні рубцювання від мостоподібного фіброзу до сформованих вузлів відбувається повне порушення архітектоніки й перехід у цироз печінки.
Показано, що фіброгенез може бути результатом невеликих, але постійних пошкоджень печінки, які призводять до репарації тканини, підтверджуючи тим самим, що активація фіброзного процесу у вигляді відкладення матриксу викликана не первинними клітинними порушеннями, а повторюваною шкідливою дією протягом певного періоду часу. Цілком імовірно, що прогресування фіброзу залежить від генетичного поліморфізму.
Чи існують варіанти розв’язання фіброзу?
Уперше вивчення матеріалу парних біопсій, взятого під час досліджень противірусних препаратів у пацієнтів із хронічними гепатитами, показало, що розпад матриксу спостерігається навіть при вираженому цирозі. Спонтанне одужання достатньої кількості експериментальних тварин із фіброзом і цирозом печінки дозволило виявити ключові моменти цього процесу [10, 11].
Відзначено, що в разі розв’язання ФП значно знижується вироблення TIMP-1 і TIMP-2, на той час як секреція матриксних металопротеїназ триває, що веде до підвищення активності колагеназ і розпаду матриксу в печінці.
Одночасно з цими змінами відбувається апоптоз ЗКП. Функція апоптозу в тканинах ссавців полягає в усуненні непотрібних клітин при їх надлишку. При прогресуючому пошкодженні печінки, коли ЗКП залучені в нормальний репаративний процес, запобігання їх апоптозу відбувається, імовірно, за допомогою сигналів розчинних факторів і змін у матриксі. Після усунення пошкоджувального агента й появи необхідності в ремоделюванні матриксу активується апоптоз ЗКП, що полегшує процес перебудови за рахунок елімінації основного джерела колагену і TIMP. Звідси логічно припустити, що вплив на процеси деградації матриксу, апоптозу ЗКП і збалансування TIMP і MMP приведе до зменшення фіброзу і повернення нормальної архітектоніки печінки. На сьогодні дослідження в цій галузі проведені тільки в експерименті.
Наведені вище клітинні й молекулярні механізми формування ФП як єдиного, послідовного процесу трансформації печінкової паренхіми дозволяють перейти до клінічних аспектів захворювання.
У світлі цього варто зупинитися на одному з найпоширеніших на сучасному етапі розвитку захворювань печінки — неалкогольній жировій хворобі печінки (НАЖХП).
Серед усіх пацієнтів із захворюваннями печінки особливий пул становлять хворі з неалкогольною жировою хворобою печінки, особливо на ранній стадії.
Поява нових неінвазивних методів діагностики дозволила скласти більш об’єктивне уявлення про клінічний перебіг захворювання й своєчасно діагностувати, а потім здійснювати моніторинг за розвитком фіброзу печінки в цієї категорії пацієнтів. Усе частіше лікарі використовують у своїй роботі еластометрію печінки для визначення стадії ФП, а також різні серологічні тести, що дозволяють діагностувати прихований фіброз [20, 21].
Прогресування фіброзу при жировій інфільтрації й за відсутності запальної активності йде не такими швидкими темпами, як при інших захворюваннях печінки (автоімунні хвороби, HCV/HBV-інфекція та інші), проте прихований розвиток процесу протягом тривалого часу призводить до істотного ФП, навіть до циротичної трансформації [22].
Більшість дослідників сходяться на думці, що саме розвиток некрозапальної реакції в гепатоцитах є ключовою стадією НАЖХП: при розвитку неалкогольного стеатогепатиту (НАСГ) темп прогресування ФП значно зростає.
Тому в даний час погляд практичних лікарів усе більше звертається в бік неінвазивних методів діагностики, поява яких дала можливість виявляти процес на ранніх стадіях, а також включити визначення стадії фіброзу в скринінг при будь-яких захворюваннях печінки, що істотно впливає на терміни та якість діагностики.
Серед неінвазивних методик виділяють фізичні (це визначення щільності печінки на апараті FibroScan) і біологічні — різні лабораторні тести [23, 24].
Слід підкреслити, що корекція способу життя є ключовою ланкою в терапії НАЖХП: доведено, що фізичне навантаження (регулярні аеробні вправи), а також зниження маси тіла зменшують ступінь стеатозу, сповільнюють прогресування ФП і значимо покращують прогноз (рівень доказовості А) [25].
Однак не для всіх пацієнтів прийнятні немедикаментозні методи, тому необхідний пошук нових способів терапевтичної корекції для лікування НАЖХП, і він активно триває. Лікарська терапія призначається при прогресуванні НАСГ, на ранній стадії процесу за наявності таких факторів ризику, як вік старше від 50 років, ЦД типу 2, метаболічний синдром, підвищений рівень АЛТ і гаммаглутамілтранспептидази [17].
Еластометрія печінки з функцією CAP і оцінка індексу фіброзу NAFLD Fibrosis Score (NFS) є універсальним інструментом як для поглибленого обстеження, так і для скринінгу в групах ризику, динамічного спостереження й оцінки ефективності терапії.
У зв’язку з цим нами проведена оцінка динаміки лабораторних тестів, що характеризують печінкове пошкодження, запалення й метаболічний профіль при НАСГ слабкої активності на тлі застосування багатофункціонального рослинного адаптогену Схізандрин®.
Cхізандрин® являє собою дієтичну добавку у формі таблетки 250 мг, до складу якої входить діюча речовина схізандрин 25 мг — комплекс біологічно активних сполук групи лігнанів лимонника китайського. Діюча речовина препарату пригнічує продукцію ФНП-α активними нейтрофілами, купферівськими клітинами й макрофагами, а також сприяє виведенню з клітин вільних радикалів. Cхізандрин® пригнічує окислювальне напруження, викликане порушенням функції мітохондрій, що запобігає некрозу й апоптозу гепатоцитів. Cхізандрин® також гальмує апоптоз гепатоцитів, стимульований ФНП-α і цитотоксичними Т-клітинами, приводячи до відновлення пошкоджень ядра й ДНК гепатоцитів.
Метою дослідження є визначення можливості «управління» процесом фіброутворення за допомогою впливу на патогенетичні механізми його формування препаратом лимонника китайського.
Обстежені 18 пацієнтів з НАСГ, середній вік становив 46,8 ± 12,2 роки. Діагноз стеатогепатита верифікувався згідно з Уніфікованим клінічним протоколом первинної, вторинної (спеціалізованної) медичної допомоги «Неалкогольний стеатогепатит», затвердженим МОЗ 06 листопада 2014 року № 826.
Концентрацію С-реактивного білка (СРБ) («Бест Діагностік», Україна), ФНП-α (АО «Вектор-Бест», Росія), інсуліну (DRG Instruments GmbH, Німеччина) натще в сироватці крові досліджували імуноферментним методом на напівавтоматичному імуноферментному мікропланшетному аналізаторі ImmunoChem-2100 (HighTechnology, Inc., США). Розраховувався індекс інсулінорезистентності (ІР) HOMA-IR за формулою: HOMA-IR = інсулін × глюкоза натще/22,5. HOMA-IR > 2,7 свідчив про наявність ІР.
Визначався індекс фіброзу NAFLD Fibrosis Score за формулою: NFS = –1,675 + 0,037 × вік (роки) + 0,094 × × індекс маси тіла ((– ІМТ (кг/м2)) + 1,13 × гіперглікемія натще/діабет (так = 1, немає = 0) + 0,99 × АСТ/АЛТ – – 0,013 × тромбоцити (× 109/л) – 0,66 × альбумін (г/дл). Рівень нижче від –1,455 свідчив про відсутність значного фіброзу, тобто F0-F2; вище від 0,676 — про наявність значного фіброзу (F3-F4). Контрольну групу становили 12 здорових осіб віком 45,3 ± 3,8 року без ознак метаболічно-асоційованих захворювань (цукровий діабет, дисліпідемія та ін.) та стеатозу печінки.
Статистичний аналіз здійснювався за допомогою програмного забезпечення Statgraphics 2.1, для порівняння показників у різних групах використовувався t-тест і тест Манна — Уїтні. Як статистично значущий розглядався p < 0,05.
Серед традиційних лабораторних показників, що характеризують функціональний стан печінки, порівняно зі здоровими особами були вірогідно підвищені рівні ліпопротеїнів низької щільності, тригліцеридів, глюкози крові, хоча останній показник не виходив за межі референсного значення, як і лужна фосфатаза, і був знижений рівень ліпопротеїнів високої щільності.
Активність АЛТ у пацієнтів із НАСГ перевищувала нормальний рівень у 2 рази, а АСТ — в 1,5 раза (табл. 2). Незначний підйом амінотрансфераз свідчив про помірно виражений некроз печінкових клітин.
/74-1.jpg)
Серед обстежених пацієнтів показник NFS вірогідно перевищував такий у контрольній групі й становив (–1,053 ± 0,962) проти (–2,879 ± 0,500); p < 0,05, що свідчило про значний процес фіброзування в печінковій тканині, незважаючи на відносно слабко виражений запальний процес, якщо його оцінювати традиційним методом за рівнем АЛТ.
Отримані дані дозволили припустити, що в обстежених пацієнтів із НАЖХП купферівські клітини перебувають у стані активності й синтезують фактор росту фібробластів, який викликає фенотипове перетворення зірчастих клітин у міофібробласти, що здійснюють синтез колагену й інших білків позаклітинного матриксу.
З огляду на значення NFS в обстежених пацієнтів мала місце загроза прогресування стеатогепатиту у фіброз і цироз печінки.
Незважаючи на те, що глікемія натще перебувала в межах референсних значень, індекс HOMA-IR у 8 разів перевищував такий у здорових осіб — (8,5 ± 2,1) проти (1,8 ± 0,9) (p < 0,05), підтверджуючи наявність вираженої ІР, яка також є фактором ризику прогресуючого перебігу НАСГ, тому що сприяє розвитку стеатозу, фіброзу й запалення (табл. 1) [19].
У пацієнтів із НАСГ відзначалося вірогідне підвищення рівня СРБ і вмісту ФНП-α порівняно зі здоровими особами — (8,3 ± 1,3) мг/л проти (2,5 ± 1,3) мг/л і (12,4 ± 1,5) пкг/мл проти (3,3 ± 1,2) пкг/мл відповідно (p < 0,01). Слід підкреслити, що ФНП-α — прозапальний цитокін який синтезується у вісцеральній жировій тканині, викликаючи місцеву, а потім і системну ІР. Він чинить плейотропну дію, стимулюючи запалення, апоптоз і відіграє ключову роль у прогресуванні НАЖХП.
Пацієнти протягом 6 місяців приймали Схізандрин® по 2 таблетки 3 рази/добу, після закінчення зазначеного строку був проведений підрахунок індексу фіброзу NFS за формулою. Виявилося, що у 88,8 % пацієнтів із НАСГ (16 осіб) через 6 місяців на тлі нормалізації метаболічних показників і печінкових проб відбувся регрес фіброзних змін у тканині печінки.
Середнє значення індексу фіброзу печінки на тлі прийому Схізандрину® становило (–2,124 ± 0,568) проти (–1,053 ± 0,962); p < 0,05. Отримані дані свідчать про здатність діючої речовини у вигляді комплексу лігнанів лимонника китайського запобігати формуванню більш тяжких стадій фіброзу печінки в пацієнтів із НАЖХП.
Вміння із сучасних позицій грамотно й своєчасно трактувати клінічні прояви захворювання печінки, розуміння механізмів їх розвитку й прогресування, знання алгоритмів ранньої діагностики допоможе клініцисту, який щодня стоїть перед вибором, визначити єдино правильну тактику ведення пацієнта із захворюванням печінки з ознаками фіброзу. Адже, як говорив Б.Є. Вотчал, «лікувати треба тоді, коли не можна не лікувати».
Конфлікт інтересів. Не заявлений.
1. Aterman K. The parasinusoidal cells of the liver: a historical account // Histochem. J. — 1986. — Vol. 18. — P. 279-305.
2. Hartroft W.S. The trabecular anatomy of late stages of experimental dietary cirrhosis. Its pathogenesis in terms of rappaport’s structural unit // The Anatomical Records. — 1954. — Vol. 119, Issue1. — P. 71-93.
3. Anthony P.P., Ishak K.G., Nayak N.C., Poulsen H.E., Scheuer P.J., Sobin L.H. The morphology of cirrhosis. Recommendations on definition, nomenclature, and classification by a working group sponsored by the World Health Organization // J. Clin Pathol. — 1978 May. — 31(5). — 395-414.
4. Boll F. Die Bindesubstanz der Drusen // Arch. Mikrosk. Anat. — 1869. — Vol. 4. — P. 334-55.
5. Kupffer K. Uber Sternzellen der Leber. Briefliche Mitteilung an Professor Waldeyer // Arch. Mikrosk. Anat. — 1876. — Vol. 12. — P. 353-8.
6. Ito T., Nemoto J. Uber die Kupfferschen Sternzellen und die ‘Fettspeicherungszellen’ (‘fat-storing cells’) in der Blutkapillarenwand der menschlichen Leber // Okajimas Folia Anat. Jpn. — 1952. — Vol. 24. — P. 243-58.
7. Wake K. Sternzellen in the liver: perisinusoidal cells with special reference to storage of vitamin A // Am. J. Anat. — 1971. — Vol. 132. — P. 429-62.
8. Reeves H.L., Burt A.D., Wood S., Day C.P. Hepatic stellate cell activation occurs in the absence of hepatitis in alcoholic liver disease and correlates with the severity of steatosis // J. Hepatol. — 1996 Nov. — 25(5). — 677-83.
9. Knook D.L., Seffelaar A.M, de Leeuw A.M. Fat-storing cells of the rat liver: Their isolation and purification // Experimental Cell Research. — 1982. — Vol. 139, Issue 2. — P. 468-471. doi: 10.1016/0014-4827(82)90283-X.
10. Yanguas S.C., Cogliati B., Willebrords J., Maes M, Colle I., van den Bossche B. et al. Experimental models of liver fibrosis // Arch. Toxicol. — 2016. — 90(5). — 1025-1048.
11. Martínez A.K., Maroni L., Marzioni M., Ahmed S.T., Milad M., Ray D. et al. Mouse models of liver fibrosis mimic human liver fibrosis of different etiologies // Curr. Pathobiol. Rep. — 2014 Dec 1. — 2(4). — 143-153.
12. Iredale J.P. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ // J. Clin. Invest. — 2007. — 117(3). — 539-548. doi.org/10.1172/JCI30542.
13. Tsuchida T., Friedman S.L. Mechanisms of hepatic stellate cell activation // Nature Reviews Gastroenterology & Hepatology. — 2017. — Vol. 14. — P. 397-411.
14. Iwaisako K., Brenner D.A., Kisseleva T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis // J. Gastroenterol. Hepatol. — 2012 Mar. — 27 (Suppl. 2). — 65-68. doi: 10.1111/j.1440-1746.2011.07002.x.
15. Higashiyama R., Inagaki Y., Hong Y.Y. et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice // Hepatology. — 2007. — Vol. 45. — P. 213-22.
16. Nallagangula K.S., Pradhan R., Shashidhar K.N. Molecular and Epigenetic Mechanisms of Bidirectional Liver Fibrosis // American J. Liver Clinical. Res. — 2017. — 1(1). — 001-006.
17. Wen Y., Jeong S., Xia Q., Kong X. Role of Osteopontin in Liver Diseases // Int. J. Biol. Sci. — 2016. — 12(9). — 1121-1128. doi: 10.7150/ijbs.16445.
18. Yanguas S.C., Cogliati B., Willebrords J., Maes M, Colle I., van den Bossche B. et al. Experimental models of liver fibrosis // Arch. Toxicol. — 2016. — 90(5). — 1025-1048.
19. Martínez A.K., Maroni L., Marzioni M., Ahmed S.T., Milad M., Ray D. et al. Mouse models of liver fibrosis mimic human liver fibrosis of different etiologies // Curr. Pathobiol. Rep. — 2014 Dec 1. — 2(4). — 143-153.
20. Perumpail B.J., Khan M.A., Yoo E.R. et al. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease // World J. Gastroenterol. — 2017. — 23(47). — 8263-76.
21. Ивашкин В.Т., Драпкина О.М., Маев И.В. и др. Распространенность неалкогольной жировой болезни печени у пациентов амбулаторно-поликлинической практики в Российской Федерации: результаты исследования DIREG 2 // Рос. журн. гастроэнтерологии, гепатологии, колопроктологии. — 2015. — 6. — 31-41.
22. Charlton M.R., Burns J.M., Pedersen R.A. et al. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States // Gastroenterology. — 2011. — 141. — 1249-53.
23. Bedossa P., Poitou C., Veyrie N. et al. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients // Hepatology. — 2012. — 56(5). — 1751-9.
24. Kanwal F., Kramer J.R., Mapakshi S. et al. Risk of Hepatocellular Cancer in Patients with Non-alcoholic Fatty Liver Disease // Gastroenterology. — 2018. — pii: S0016-5085(18)34889-3.
25. Wang K. Molecular mechanisms of hepatic apoptosis // Cell Death Dis. — 2014. — 5. — e996.