Синдром Луї-Бар — рідкісне генетичне захворювання, що пов’язано з мутаціями в гені ATM (Ataxia Telangiectasia Mutated) і проявляється у вигляді характерної тріади синдромів: атаксія, телеангіектазії, імунодефіцит [1, 2, 4]. Має автосомно-рецесивне спадкування. Частота синдрому у світі становить 1 випадок на 40–100 тисяч дітей. Належить до групи хвороб з нестабільністю геному і порушеннями процесів репарації ДНК. Мозочкова атаксія при синдромі Луї-Бар маніфестує зазвичай з кінця першого року життя і може бути початковим клінічним проявом хвороби [10]. Телеангіектазії на шкірі і слизових оболонках іноді бувають поодинокими, тому часто випадають з поля зору клініцистів або розвиваються пізніше, у віці 3–5 років, коли пацієнт вже тривалий час спостерігається у неврологів з приводу атаксії нез’ясованого походження або дитячого церебрального паралічу [16]. Комбінований імунодефіцит при синдромі Луї-Бар також може мати відтерміновану клінічну маніфестацію, у зв’язку з чим мозочкова атаксія нерідко є моносиндромом протягом певного часу [10]. Тому вперше дитина нерідко потрапляє під диспансерний нагляд саме дитячого невролога з симптомами прогресуючої мозочкової атаксії, а не до клінічного імунолога з картиною частих інфекцій та/або імунозалежних проявів. Хоча найтяжчі ускладнення у дітей з синдромом Луї-Бар часом пов’язані саме з імунодефіцитом, а не з ураженням ЦНС, і полягають у розвитку тяжких, загрозливих для життя інфекцій, зумовлених опортуністичними вірусами [8, 13] і грибками [9], та низки автоімунних хвороб [5, 14, 15] і злоякісних новоутворень [8, 13, 17]. S. Аlyasin зі співавт. показали, що серед клінічних симптомів синдрому Луї-Бар у когорті досліджуваних пацієнтів переважали атаксія (100 %), окулокутанні телеангіектазії (77,8 %) дизартрія й окорухова апраксія (72,2 %), рецидивні інфекції (70,6 %) та гостра лімфобластна лейкемія (16,7 % випадків) [6].
У зв’язку з цим неврологи мають бути добре поінформовані щодо цієї форми первинного імунодефіциту, щоб своєчасно направляти пацієнтів до клінічних імунологів для проведення імунологічних досліджень і генетичної верифікації діагнозу [2].
При імунологічному дослідженні зазвичай відзначається дефіцит клітинної ланки імунітету, а саме Т-лімфоцитів, Т-хелперів і цитотоксичних Т-клітин. Серед гуморальних порушень переважає вибірковий дефіцит IgA. За даними S. Аlyasin зі співавт., при дослідженні імунного статусу в когорті пацієнтів з синдромом Луї-Бар відзначалась знижена сироваткова концентрація молекул IgG (33,3 %) та IgA (40,0 %), а також мала кількість B-лімфоцитів (46,67 %) і T-клітин (73,33 %) у крові [6].
Характерним для синдрому Луї-Бар є підвищення сироваткової концентрації альфа-фетопротеїну, що може бути причиною помилкового пошуку гепатоцелюлярної карциноми [2].
Діти з синдромом Луї-Бар патологічно високочутливі до іонізуючої радіації через нестабільність ДНК, тому для них можуть бути шкідливими рентгенологічні обстеження, а проведення променевої терапії при злоякісних новоутвореннях нерідко є фатальним [7].
У даній публікації наведено опис історії хвороби дитини з верифікованим діагнозом синдрому Луї-Бар із власної клінічної практики, щоб привернути увагу неврологів до цієї форми первинного імунодефіциту з неврологічними проявами.
Пацієнтка — дівчинка віком 2 роки 6 місяців. Батьки дитини звернулися на прийом до нейроімунолога зі скаргами на хиткість і низьку витривалість при ходьбі, періодичні падіння в дитини. Також відзначалися періодичні спазми жувальних м’язів.
З анамнезу з’ясували, що дитина народилася доношеною після фізіологічних пологів. Перебіг вагітності був неускладненим. Протягом перших місяців життя не відзначалося патологічних відхилень у стані здоров’я. Згодом відзначили, що дитина пізно почала ходити — лише у віці 1 рік і 4 місяці. Хода була невпевненою, дитина швидко виснажувалася. З часом не відзначалося поліпшення навиків ходьби. Мала місце виражена хиткість при пересуванні, так звана п’яна хода. Пізніше приєдналися періодичні напади спазмів жувальних м’язів, що призводили до скреготу зубів. Після досягнення віку 2 років почали з’являтися рецидивні виразки на слизовій оболонці ротової порожнини невеликого розміру, оточені червоною облямівкою. Тоді ж помітили збільшення підщелепних і шийних лімфатичних вузлів.
При фізикальному огляді шкірні покриви блідо-рожевого кольору, чисті. Ретельний огляд як шкіри, так і доступних слизових оболонок не виявив ознак телеангіектазій. Відзначається збільшення піднебінних мигдаликів 2-го ступеня. Пальпаторно підщелепні, передньо- і задньошийні лімфовузли помірно збільшені, неболючі, рухливі. В легенях вислуховується симетричне везикулярне дихання з обох боків. Серцеві тони ритмічні, звучні, чисті. При пальпації живіт м’який, безболісний у всіх відділах. Симптом Пастернацького негативний з обох боків. Фізіологічні відправлення в нормі.
При неврологічному огляді свідомість дівчинки ясна. Психічно розвинена згідно з віком. З боку черепно-мозкових нервів порушення фіксації зору на статичному та рухомому предметі при відсутності ознак паралічу нервів, що рухають очні яблука, — ознаки окорухової апраксії. Відзначаються рідкісні поодинокі неболючі спазми жувальних м’язів з обох боків. Розладів чутливості не зареєстровано. Сухожилкові та періостальні рефлекси дифузно пожвавлені з обох боків, викликаються з розширених рефлексогенних зон. Патологічних стопних і кистьових рефлексів не відзначається. Координаторні проби виконує з помірним мимопопаданням та інтенційним тремором. Відзначається дисметрія й адіадохокінез. Має місце дифузна гіпотонія м’язів кінцівок. Сила м’язів в руках — 5 балів, в ногах — 4,5 бала. В простій позі Ромберга виражена хиткість із тенденцією до падіння назад. В ускладненій позі Ромберга нестійка. Хода з вираженим похитуванням у боки з тенденцією до падіння на поворотах. Функція тазових органів не порушена.
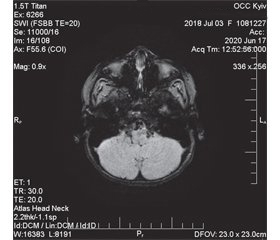
Таким чином, у дитини відзначили прояви статичної та динамічної мозочкової атаксії, окорухової апраксії та екстрапірамідний синдром у вигляді бруксизму. Дані історії хвороби вказують, що атаксія була природженою на тлі сприятливого анте- та перинатального анамнезу. Як відомо, синдром Луї-Бар є важливою причиною природженої мозочкової атаксії у дітей, і його діагноз має обов’язково розглядатися при проведенні диференціальної діагностики з ознаками дисфункції мозочка. Наявність окорухової апраксії та м’язової дистонії була додатковим аргументом на користь діагнозу синдрому Луї-Бар, оскільки ці клінічні прояви є характерними для таких дітей. МРТ головного мозку в конвенційних режимах без контрасту демонструвала початкові ознаки атрофії мозочка (рис. 1А) та патологічне майже симетричне розширення задніх рогів бічних шлуночків півкуль великого мозку (рис. 1Б). Ці нейровізуалізаційні дані відповідають результатам дослідження R.A. Dineen зі співавт. [10].
/777.jpg)
З метою підтвердження клінічного діагнозу були призначені спеціальні генетичні тести для визначення стану гена АТМ, імунологічне обстеження для пошуку ознак типового для синдрому Луї-Бар комбінованого імунодефіциту та вимірювання сироваткової концентрації альфа-фетопротеїну, що є загальновизнаним інформативним лабораторним біомаркером при цій хворобі.
Генетичний тест, проведений в діагностичному центрі Сentogene (Росток, Німеччина), виявив три мутації в гені АТМ, зокрема дві відомі патогенні заміни нуклеотидів у гетерозиготному стані — с.8147Т>С (p.Val2716Ala) і c.8584+2T>C (Splice donor) та одну невідому раніше мутацію — c.3178A>G (p.fle1060Val) — невизначеного діагностичного значення в гетерозиготному стані. Ці дані верифікували клінічний діагноз синдрому Луї-Бар як генетичної хвороби в обстежуваної пацієнтки.
Імунологічне дослідження включало вивчення показників загального аналізу крові, субпопуляційного складу лімфоцитів із застосуванням лазерної проточної цитофлуориметрії (цитофлуориметр Epics Xl, США) і методу непрямої імунофлуоресценції з моноклональними антитілами до CD-маркерів з двома або трьома мітками (CD3+, CD3+CD4+, CD3+CD8+, CD3–CD19+, CD3–CD16+CD56+, CD3+CD16+CD56+) (реактиви Beckman Coulter, США). Фагоцитоз оцінювали за даними латекс-тесту з визначенням показника фагоцитозу, фагоцитарного індексу, кількості активних фагоцитів і фагоцитарної ємності крові. Сироваткові концентрації імуноглобулінів основних класів (М, G, А) визначали за результатами простої радіальної імунодифузії за Манчіні. Концентрацію класів IgE, IgD та субкласів IgG (IgG1, IgG2, IgG3, IgG4) у сироватці крові вимірювали за допомогою імуноферментного аналізу («Вектор-Бест», РФ). Активність мієлопероксидази нейтрофілів оцінювали імуноферментним методом в лабораторії ней-роімунології Інституту нейрохірургії імені А.П. Ромоданова. Там же проводили НСТ-тест. Концентрацію манозозв’язуючого лектину в сироватці крові визначали методом ELISA в Німеччині за допомогою лабораторії доктора Рьодгера.
В результаті проведеного імунологічного дослідження ідентифікували ознаки комбінованого імунодефіциту, типового для синдрому Луї-Бар, а саме лімфопенію, дефіцит CD3+ Т-лімфоцитів (49 % при нормі 54–80 %), CD3+CD4+ Т-хелперів (24 % при нормі 31–49 %), CD3+CD8+ цитотоксичних Т-клітин (13 % при нормі 16–38 %) та вибірковий дефіцит IgA (сироваткова концентрація 0,31 г/л при нормі 0,6–2,5 г/л) за нормальних сироваткових концентрацій імуноглобулінів інших класів. Ультразвукове дослідження імунних органів та органів черевної порожнини продемонструвало ознаки акцидентальної трансформації тимуса, лімфаденопатії шийних і підщелепних лімфатичних вузлів, помірного гепатолієнального синдрому.
З метою пошуку причини лімфопроліферативних змін (гіпертрофії піднебінних мигдаликів, лімфаденопатії, акцидентальної трансформації тимуса, гепатоспленомегалії) проведено ПЛР лейкоцитів крові з видоспецифічними праймерами HSV1/2, VZV, EBV, CMV, HHV-6. HHV-7, HHV-8, аденовірусів, ентеровірусів, TTV, парвовірусу В19, вірусів гепатиту В, С, D і G, T. gondii, Borrelia burg., Chlamydia pneum., Mуcoplasma pneum. в лабораторії нейробіохімії Інституту нейрохірургії імені А.П. Ромоданова. За результатами цих досліджень ідентифікували ДНК EBV у великій кількості — більше 100 тисяч вірусних частинок у пробі.
Відзначалася значно підвищена сироваткова концентрація лабораторного біомаркера синдрому Луї-Бар — білка альфа-фетопротеїну, що зміцнювало переконаність у правильності клінічного діагнозу. Її рівень на момент обстеження становив 30,6 МО/мл при нормі менше 2,64 МО/мл.
Таким чином, клінічний діагноз синдрому Луї-Бар у цьому випадку не викликав сумнівів. Відзначалися типові ознаки хвороби, зокрема природжена прогресуюча мозочкова атаксія та комбінований імунодефіцит з характерним патерном імунологічних порушень. Мало місце підвищення концентрації альфа-фетопротеїну у сироватці крові. Були ідентифіковані відповіді мутації в гені АТМ, що лежать в основі цього синдрому. Відсутність характерних телеангіектазій можна пояснити віком пацієнтки на момент обстеження. Як відомо, при синдромі Луї-Бар цей клінічний феномен може з’являтися дещо пізніше, у віці 3–5 років.
Клінічний діагноз: синдром Луї-Бар (с.8147Т>С (p.Val2716Ala), c.8584+2T>C (Splice donor), c.3178A>G (p.fle1060Val)) — неврологічний синдром у вигляді мозочкової атаксії, окорухової апраксії, бруксизму та комбінований імунодефіцит (дефіцит Т-лімфоцитів, Т-хелперів, цитотоксичних Т-клітин та IgA).
Відповідно до отриманих клініко-лабораторних даних було призначено комплексне лікування. Для пригнічення репродуктивної активності герпесвірусної інфекції призначили противірусний хіміопрепарат валганцикловір перорально в дозі 450 мг двічі на добу протягом 1 місяця, за рахунок чого вдалося отримати негативний результат ПЛР крові і зменшити прояви афтозного стоматиту, лімфаденопатії та гепатолієнального синдрому в кінці курсу терапії. Для компенсації комбінованого імунодефіциту був призначений препарат діалізату суспензії лейкоцитів крові в дозі 4 мл в/м 1 раз на тиждень № 5. Раніше повідомляли про успіхи при застосуванні цього імунотерапевтичного агента при первинних клітинних і комбінованих імунодефіцитах людини [3], включаючи синдроми Віскотта — Олдрича [12] та Челдіака — Хігаші [11]. Для підвищення ефекту діалізату лейкоцитів крові додатково призначили пропес (екстракт альфа- і бета-дефензинів) у дозі 2 мл в/м через день на ніч № 15. За рахунок цієї імунотерапії вдалося досягти позитивних змін з боку порушених імунологічних показників — відзначалося зростання кількості Т-хелперів та цитотоксичних Т-лімфоцитів у крові, що вказувало на часткову компенсацію наявного імунодефіциту. З метою лікування мозочкової атаксії призначили нейротропні препарати цереброкурин у дозі 2 мл в/м зранку через день № 15 та цитиколін у дозі 1000 мг (4 мл) в/м через день зранку № 15, чергуючи з цереброкурином, і отримали невеликий позитивний ефект в кінці курсу терапії, що полягав у деякому покращенні стійкості в позі Ромберга та утриманні рівноваги при ходьбі.
Даний клінічний приклад яскраво демонструє потенціал нейроімунологічного підходу до ведення пацієнтів, оскільки досліджувана дитина страждала як від імунодефіциту, так і від неврологічної дисфункції, що пов’язано з плейотропними ефектами мутованого гена, який був причиною розвитку хвороби. Отже, пацієнтка мала бути під наглядом щонайменше двох різних медичних спеціалістів — клінічного імунолога та невролога. Аналогічно з цим продемонстрована користь від подвійного підходу до терапії в таких випадках — імунотерапії та лікування опортуністичної інфекції за імунологічним напрямком та ноотропної неврологічної терапії і нейрореабілітації відповідно до симптомів ураження ЦНС. Реактивовані форми EBV-інфекції дуже характерні для синдрому Луї-Бар [18, 19]. Хоча в даному клінічному випадку йдеться про доброякісну вірус-індуковану лімфопроліферацію, в науковій літературі існує чимало повідомлень про злоякісні пухлини, викликані EBV, при цій формі первинного імунодефіциту, включаючи MALT-лімфому [8] та хворобу Ходжкіна [13]. Своєчасне виявлення і адекватне лікування реактивованої опортуністичної вірусної інфекції може бути запорукою профілактики низки злоякісних новоутворень у дітей з синдромом Луї-Бар. Показано також позитивний вплив препарату діалізату лейкоцитів крові на стан клітинного імунітету у цих дітей, що може бути використано при проведенні подальших клінічних випробувань імунотерапевтичних підходів у імуноскомпрометованих пацієнтів.
Конфлікт інтересів. Автор заявляє про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Список литературы
1. Евтушенко С.К. и др. Неврология раннего детского возраста. Киев, 2016. 287 с.
2. Мальцев Д.В., Казмирчук В.Е. Иммунодефицитные болезни человека. Киев: Феникс, 2012. 596 с.
3. Мальцев Д.В. Показания к применению трансфер факторов в клинической практике. Імунологія і алергологія: наука і практика. 2019. № 2. С. 4-20.
4. Мухин А.С. Неврология детского возраста. Москва: Медицина, 2004. 764 с.
5. Abdulhag U.N., Liebster D., Eisenstein E.M., Berkun Y. Efficacy of Rituximab in Refractory Cold Agglutinin Hemolytic Anemia in a Patient with Ataxia-Telangiectasia. Isr. Med. Assoc. J. 2015. Vol. 17(7). P. 455-456.
6. Alyasin S., Esmaeilzadeh H., Ebrahimi N. et al. Clinical Presentation of Ataxia-Telangiectasia. Arch. Iran. Med. 2019. Vol. 22(12). P. 682-686.
7. Asadollahi R., Britschgi C., Joset P. et al. Severe reaction to radiotherapy provoked by hypomorphic germline mutations in ATM (ataxia-telangiectasia mutated gene). Mol. Genet. Genomic. Med. 2020. e1409.
8. Bennett J.A., Bayerl M.G. Epstein-Barr virus-associated extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT Lymphoma) arising in the parotid gland of a child with ataxia telangiectasia. J. Pediatr. Hematol. Oncol. 2015. Vol. 37(2). e114-117.
9. Cipe F., Dogu F., Yildiran A., Yüksek M. An unusual clinical presentation: invasive Candida non-albicans infections in ataxia telangiectasia. J. Investig. Allergol. Clin. Immunol. 2008. Vol. 18(6). P. 488-490.
10. Dineen R.A., Raschke F., McGlashan H.L., Pszczolkowski S. et al. Multiparametric cerebellar imaging and clinical phenotype in childhood ataxia telangiectasia. Neuroimage Clin. 2020. Vol. 25. P. 102-110.
11. Khan A., Hill J.M., Loeb E., MacLellan A., Hill N.O. Mana-gement of Chédiak-Higashi syndrome with transfer factor. Am. J. Dis. Child. 1973. Vol. 126(6). P. 797-799.
12. Levin A.S., Spitler L.E., Stites D.P., Fudenberg H.H. Wiskott-Aldrich syndrome, a genetically determined cellular immunologic deficiency: clinical and laboratory responses to therapy with transfer factor. Proc. Natl. Acad. Sci. U S A. 1970. Vol. 67(2). P. 821-828.
13. Li X.L., Wang Y.L. Ataxia-telangiectasia complicated with Hodgkin’s lymphoma: A case report. World. J. Clin. Cases. 2020. Vol. 8(11). P. 2387-2391.
14. Pasini A.M., Gagro A., Roić G. et al. Ataxia Telangiectasia and Juvenile Idiopathic Arthritis. Pediatrics. 2017. Vol. 139(2). e20161279.
15. Patiroglu T., Gungor H.E., Unal E. et al. Hashimoto thyroiditis associated with ataxia telangiectasia. J. Pediatr. Endocrinol. Metab. 2012. Vol. 25(3–4). P. 349-352.
16. Perez Maturo J., Gonzalez Cid M., Zavala L. et al. Novel Variants in ATM Causing Mild Ataxia-Telangiectasia: From Benchside to Bedside and Back Again. Mov. Disord. Clin. Pract. 2020. Vol. 7(6). P. Р. 727-729.
17. Pitter K.L., Casey D.L., Lu Y.C. et al. Pathogenic ATM Mutations in Cancer and a Genetic Basis for Radiotherapeutic Efficacy. J. Natl. Cancer Inst. 2020. djaa095.
18. Rubinstein J.D., Burns K., Absalon M. et al. EBV-directed viral-specific T-lymphocyte therapy for the treatment of EBV-driven lymphoma in two patients with primary immunodeficiency and DNA repair defects. Pediatr. Blood Cancer. 2020. Vol. 67(3). e28126.
19. Tatfi M., Hermine O., Suarez F. Epstein-Barr Virus (EBV)-Related Lymphoproliferative Disorders in Ataxia Telangiectasia: Does ATM Regulate EBV Life Cycle? Front. Immunol. 2019. Vol. 9. P. 3060.

/777.jpg)