Резюме
Актуальність. Гіперфенілаланінемія (HPA), або підвищення концентрації фенілаланіну (Phe) в крові, є наслідком порушення перетворення фенілаланіну в тирозин. Яскравим прикладом цього клінічного стану є класична фенілкетонурія (PKU). У 1970-х роках стало зрозуміло, що в частини хворих із PKU, незважаючи на ефективну дієтотерапію, розвивалися тяжкі неврологічні порушення. Ці випадки верифікували як атипові, або злоякісні, варіанти PKU, що, як було досліджено, спричинялось мутаціями в генах, які регулюють синтез та регенерацію тетрагідробіоптерину (ВН4). Порушення метаболізму тетрагідробіоптерину, або ВН4-дефіцитні синдроми, — гетерогенна група спадкових захворювань ЦНС, спричинених дефіцитом моноамінів-нейротрансмітерів, які в низці випадків супроводжуються гіперфенілаланінемією. ВН4-дефіцитні синдроми, які є рідкісними вродженими порушеннями метаболізму, спричиненими недостатністю нейротрансмітерів, за відсутності своєчасного лікування призводять до прогресуючої, нерідко летальної, енцефалопатії. Упровадження в медичну практику ранньої диференціальної клінічної та лабораторної діагностики гіперфенілаланінемій дозволить запобігти розвитку летальних енцефалопатій та суттєво підвищити ефективність їх лікування. Матеріали та методи. Були обстежені 59 пацієнтів із діагнозом PKU віком від 2 до 16 років. Гіперфенілаланінемія була загальним симптомом і встановлена в усіх обстежених пацієнтів. Усім хворим було проведено молекулярно-генетичне обстеження на панель мутацій у гені РАН (R408W, Y414C (12-й екзон), R158Q (5-й екзон), Ivs10nt546 (5-й інтрон), Ivs12nt1 (12-й інтрон), R252W, R261W, R261Q, R261X, G272X, P281L, S273F (7-й екзон). Усі хворі були обстежені на спектр птеринів (біоптерин та неоптерин) в крові та сечі. У 4 пацієнтів із вираженими неврологічними симптомами додатково визначали активність дигідроптеринредуктази в сухих краплях крові. Двом пацієнтам проводилося вимірювання концентрації гомованілової кислоти та 5-гідроксііндолоцтової кислоти в спинномозковій рідині методом високоефективної рідинної хроматографії. Усі пацієнти лікувались за допомогою дієтотерапії з використанням продуктів спеціалізованого лікувального харчування. Результати. 59 пацієнтів із гіперфенілаланінемією були обстежені на птериновий профіль. У 55 пацієнтів був установлений діагноз PKU, у 4 пацієнтів із прогресуючим ураженням нервової системи та порушенням птеринового профілю було встановлено діагноз ВН4-дефіцитних синдромів: 3 пацієнти — із діагнозом «PTPS-синдром», 1 пацієнт — «DHPR-синдром». Основні клінічні прояви ВН4-дефіцитних синдромів у виявлених пацієнтів були такими: затримка розумового розвитку, судоми, прогресування екстрапірамідних симптомів, порушення тонусу м’язів і пози, ковтання, гіперсалівація. Перші симптоми виникали частіше протягом перших місяців життя дитини. Головним біохімічним маркером на початку захворювання була гіперфенілаланінемія. При застосуванні дієтотерапії рівень Phe швидко досягав рекомендованих безпечних для нервової системи значень, але хвороба прогресувала. Висновки. Гіперфенілаланінемія — основний прояв гетерогенної групи рідкісних нейрометаболічних захворювань, які вимагають подальшої діагностичної верифікації. У новонароджених, у яких під час проведення масового неонатального скринінгу на фенілкетонурію виявлений підвищений рівень фенілаланіну, необхідно застосовувати додатковий діагностичний протокол (визначення птеринового профілю в крові та сечі, обов’язкове дослідження активності дигідроптеринредуктази у сухій краплі крові). Дослідження птеринів необхідне в пацієнтів із раніше встановленим діагнозом «фенілкетонурія» за неефективності дієтотерапії, прогресування когнітивних і неврологічних порушень, зниження тонусу м’язів, виникнення судомного синдрому, наявності екстрапірамідних симптомів. Рання діагностика ВН4-дефіцитних синдромів та своєчасно розпочате лікування дозволяють суттєво покращити стан хворих. Для деяких ВН4-дефіцитних синдромів гіперфенілаланінемія не характерна, що унеможливлює раннє виявлення таких пацієнтів під час неонатального скринінгу. Тому виявлення прогресуючих неврологічних симптомів у пацієнтів віком до 1 року й у ранньому дитинстві, які супроводжуються втратою когнітивних функцій на тлі м’язової гіпотонії, дистонічних порушень і судом, зумовлює необхідність дослідження птеринового профілю в крові й сечі.
Актуальность. Гиперфенилаланинемия (HPA), или повышение концентрации фенилаланина в крови (Phe), является следствием нарушения превращения фенилаланина в тирозин. Классическим примером HPA является классическая фенилкетонурия (PKU). В 1970-х годах стало понятно, что у части больных с PKU независимо от эффективности проводимой диетотерапии развивались тяжелые неврологические нарушения. Эти случаи верифицировали как атипичную, или злокачественную, PKU. Эти атипичные варианты PKU вызваются мутациями в генах, регулирующих синтез и регенерацию тетрагидробиоптерина (ВН4). Нарушение метаболизма тетрагидробиоптерина, или ВН4-дефицитные синдромы, — гетерогенная группа наследственных заболеваний ЦНС, обусловленных дефицитом моноаминов-нейротрансмиттеров, которые в ряде случаев сопровождаются гиперфенилаланинемией. ВН4-дефицитные синдромы относятся к редким нарушениям метаболизма, и отсутствие своевременного лечения приводит к развитию летальных энцефалопатий. Внедрение в медицинскую практику ранней дифференциальной клинической и лабораторной диагностики гиперфенилаланинемий позволит существенно повысить эффективность лечения этих фатальных нейрометаболических синдромов. Материалы и методы. Были обследованы 59 пациентов с диагнозом PKU в возрасте от 2 до 16 лет. Гиперфенилаланинемия была общим симптомом и диагностирована у всех обследованных пациентов. Всем больным было проведено молекулярно-генетическое обследование на панель мутаций в гене РАН (R408W, Y414C (12-й экзон), R158Q (5-й экзон), Ivs10nt546 (5-й интрон), Ivs12nt1 (12-й интрон), R252W, R261W, R261Q, R261X, G272X, P281L, S273F (7-й экзон). Все больные были обследованы на спектр птеринов (биоптерин и неоптерин) в крови и моче. У 4 пациентов с выраженными неврологическими симптомами дополнительно определяли активность дигидроптеринредуктазы в сухих каплях крови. Двум пациентам проводилось измерение концентрации гомованиловой и 5-гидроксииндолуксусной кислоты в спинномозговой жидкости методом высокоэффективной жидкостной хроматографии. В лечении всех пациентов использовали диетотерапию, они принимали продукты специализированного лечебного питания. Результаты. 59 пациентов с гиперфенилаланинемией были обследованы на птериновый профиль. У 55 пациентов была диагностирована PKU, у 4 пациентов с прогрессирующим поражением нервной системы и нарушениями птеринового профиля был установлен диагноз ВН4-дефицитных синдромов: 3 пациента — с диагнозом «PTPS-синдром», 1 пациент — «DHPR-синдром». Основными клиническими проявлениями ВН4-дефицитных синдромов у выявленных пациентов были: задержка умственного развития, судороги, прогрессирование экстрапирамидных симптомов, нарушения тонуса и позы, глотания и гиперсаливация. Первые симптомы возникали чаще на протяжении первых месяцев жизни ребенка. Главным биохимическим маркером в начале заболевания была гиперфенилаланинемия. На фоне применяемой диетотерапии уровень Phe быстро достигал рекомендованных безопасных для нервной системы значений, но болезнь все равно прогрессировала. Выводы. Гиперфенилаланинемия — основной симптом гетерогенной группы редких нейрометаболических заболеваний, которые требуют дальнейшей диагностической верификации. У новорожденных, у которых во время проведения массового неонатального скрининга на фенилкетонурию, выявлены высокие уровни фенилаланина, необходимо использовать дополнительный диагностический протокол (определение птеринового профиля в крови и моче, исследование активности дигидроптеринредуктазы в сухой капле крови). Исследование птеринов необходимо у пациентов с диагнозом «фенилкетонурия» при неэффективности диетотерапии, прогрессировании когнитивных и неврологических нарушений, снижении мышечного тонуса, эпилептическом синдроме и экстрапирамидных нарушениях. Ранняя диагностика ВН4-дефицитных синдромов и своевременное лечение позволяют улучшить состояние больных. Для некоторых ВН4-дефицитных синдромов гиперфенилаланинемия не характерна, что не позволяет выявлять этих пациентов во время неонатального скрининга. Поэтому выявление прогрессирующих неврологических симптомов у пациентов в возрасте до 1 года и в раннем детстве, которые сопровождаются утратой когнитивных навыков, мышечной гипотонией, дистоническими нарушениями, эписиндромом, требует исследования птеринового профиля в крови и моче.
Background. Hyperphenylalaninemia, or increased concentration of phenylalanine in the blood, is a consequence of a disorder of phenylalanine conversion to tyrosine. A classic example of hyperphenylalaninemia is phenylketonuria (PKU). In the 1970s, it became clear that some neurological disorders developed in part of patients with PKU, regardless of the effectiveness of dietary therapy. These cases were verified as atypical, or malignant, PKU. These atypical PKU variants are caused by mutations in the genes that regulate tetrahydrobiopterin (BH4) synthesis and regeneration. Disorders of tetrahydrobiopterin metabolism, or BH4 deficiencies, are a heterogeneous group of hereditary diseases of the central nervous system caused by a deficiency of monoamine neurotransmitters, which in some cases are accompanied by hyperphenylalaninemia. BH4 deficiencies are the rare metabolic disorders, and the lack of timely treatment leads to the development of lethal encephalopathies. The introduction into clinical practice of early differential clinical and laboratory diagnosis of hyperphenylalaninemia will significantly improve the effectiveness of treatment of these fatal neurometabolic syndromes. Materials and methods. We examined 59 patients diagnosed with PKU aged 2 to 16 years. Hyperphenylalaninemia was a common symptom and was diagnosed in all the patients. All patients underwent a molecular genetic examination on the panel of mutations in the phenylalanine hydroxylase gene (R408W, Y414C (exon12), R158Q (exon 5), Ivs10nt546 (intron 5), Ivs12nt1 (intron 12), R252W, R261W, R261Q, R261X, G272X, P281L, S273F (exon 7). Also, all patients were examined on a spectrum of pterins (biopterin and neopterin) in the blood and urine. In 4 patients with severe neurologic symptoms, the activity of dihydropterin reductase in dry blood spots was additionally determined. Two patients were measured the concentration of homovanillic and 5-hydroxyindoleacetic acids in cerebrospinal fluid by means of high-performance liquid chromatography. All patients used diet therapy and received specialized therapeutic nutrition. Results. Fifty-nine patients with hyperphenylalaninemia were examined for the pterin profile. In 55 patients, PKU was diagnosed, in 4 patients with progressive neurological symptoms and abnormalities of pterin profile, BH4 deficiencies were diagnosed: in 3 patients — with 6-pyruvoyltetrahydropterin synthase and in 1 patient — with dihydropteridine reductase deficiency. The main clinical manifestations of BH4 deficiency in the identified patients were: mental retardation, convulsions, progression of extrapyramidal symptoms, tonus and posture disorders, impaired swallowing and hypersalivation. First symptoms occurred more often during the first months of the child’s life. The main biochemical marker at the beginning of the disease was hyperphenylalaninemia. Against the background of dietary therapy, phenylalanine level quickly reached the recommended values safe for the nervous system, but the disease still progressed. Conclusions. Hyperphenylalaninemia is the main symptom of a heterogeneous group of rare neurometabolic diseases that require further diagnostic verification. In newborns, who have high levels of phenylalanine at the time of mass neonatal screening for phenylketonuria, an additional diagnostic protocol (the determination of pterin levels in the blood and urine, the activity of dihydropterin reductase in dry blood spot) should be used. The examination of pterin profile is necessary in patients diagnosed with phenylketonuria and failure of the diet therapy, progression of cognitive and neurological disorders, decreased muscle tone, epileptic syndrome and extrapyramidal disorders. The early diagnosis of BH4 deficiency and timely treatment can improve the clinical condition of the patients. The hyperphenylalaninemia is not characteristic for some BH4 deficiencies, it makes impossible to identify such patients during mass neonatal screening. Therefore, the detection of progressive neurological symptoms in these patients under the age of 1 year and in early childhood, which are accompanied by a loss of cognitive skills, muscle hypotension, dystonic disorders, epileptic syndrome, requires the necessary study of pterin profile in the blood and urine.
Вступ
Діагностика орфанних метаболічних захворювань стала можливою завдяки проведенню масового неонатального скринінгу (МНС). Під час проведення МНС виявляється підвищений рівень фенілаланіну (Phe), або гіперфенілаланінемія (HPA). Підвищення концентрації фенілаланіну в крові є наслідком порушення перетворення фенілаланіну в тирозин. До недавнього часу вважали, що HPA є ознакою тільки одного захворювання, відомого як фенілкетонурія (PKU). Але дослідження метаболізму Phe показали, що гіперфенілаланінемія — це основна біохімічна ознака цілої групи орфанних нейрометаболічних захворювань, яку доречно визначають як гіперфенілаланінемії. Нозологічні форми гіперфенілаланінемії потребують верифікації та застосування різних підходів до лікування.
PKU є найбільш поширеним захворюванням серед гіперфенілаланінемій. Як відомо, основними симптомами класичної фенілкетонурії є висока концентрація Phe в крові та проміжних речовин його метаболізму (особливо фенілпірувату, фенілацетату та феніллактату) в сечі і розвиток тяжкої розумової недостатності у випадках несвоєчасно розпочатого лікування [1]. Класична PKU є результатом зниженої активності фенілаланінгідроксилази (PAH), що знаходиться виключно в печінці у людини. Застосування спеціальної дієти з використанням лікувальних амінокислотних сумішей із низькім умістом фенілаланіну та підвищеним рівнем тирозину з місячного віку запобігає розвитку розумової відсталості в пацієнтів з PKU. У 1970-х роках стало зрозуміло, що у частини хворих із PKU, незважаючи на ефективну дієтотерапію, розвивались тяжкі неврологічні порушення. Ці випадки верифікували як атипові (або злоякісні) варіанти PKU, що, як було досліджено, спричинялося мутаціями в генах, які регулюють синтез та регенерацію тетрагідробіоптерину (ВН4) [2]. Атипові форми PKU становлять 1–3 % серед пацієнтів із HPA і зустрічаються з частотою 1 : 500 000–1 : 100 000 000 новонароджених. Завдяки вдосконаленню хімічного синтезу було отримано лімітовану кількість синтетичних біоптеринових сполук, призначення яких хворим з атиповою формою PKU приводило до швидкої нормалізації рівня Phe незалежно від застосування спеціалізованої дієти [3]. Таким чином, були описані інші нозологічні форми гіперфенілаланінемій, пов’язані з метаболізмом ВН4.
ВН4 — основний кофактор трьох гідроксилаз (фенілаланінгідроксилази, тирозингідроксилази, триптофангідроксилази), необхідних для синтезу моноамінових нейротрансмітерів, зокрема дофаміну, адреналіну (епінефрину), норадреналіну (норепінефрину), серотоніну, мелатоніну [4]. Крім того, BH4 — основний кофактор трьох ізоформ синтетази оксиду азоту (NOS), порушення структури яких спричиняє виникнення кардіоваскулярної та ендотеліальної дисфункції і, ймовірно, зміну больової модуляції [5]. Таким чином, спадковий дефіцит ферментів, що беруть участь у синтезі та подальшому метаболізмі BH4, спричиняє порушення активності трьох гідроксилаз (фенілаланінгідроксилази, тирозингідроксилази, триптофангідроксилази) і суттєві порушення метаболізму біогенних амінів.
Порушення метаболізму тетрагідробіоптерину, або ВН4-дефіцитні синдроми, — гетерогенна група спадкових захворювань ЦНС, спричинених дефіцитом моноамінів-нейротрансмітерів, які в низці випадків супроводжуються гіперфенілаланінемією. Патогенетичні біохімічні ланки нейрометаболічних порушень при ВН4-дефіцитних станах локалізовані у структурах ЦНС і печінці (ураження периферичної печінкової фенілаланінгідроксилазної системи) [6].
Основна причина виникнення ВН4-дефіцитних станів — мутація генів, які кодують ферменти біосинтезу та регенерації ВН4. До ферментів біосинтезу належать GTP циклогідроксилаза, 1 GTPCH і 6-пірувол-тетрагідроптеринсинтаза (PTPS); до ферментів регенерації — птерин-4α-кабіноламіндегідратаза (PCD)/димеризаційний кофактор нуклеарного фактора гепатоцитів 1α PCD/DCoH та дигідроптеринредуктаза (DHPR) [7].
Для ВН4-дефіцитних синдромів характерні різноманітні клінічні фенотипи зі значними варіаціями дебюту захворювання та його проявів, що зумовлене місцезнаходженням дефектного ферменту в метаболічному циклі ВН4, а також варіантом мутації та типом успадкування [8]. Автосомно-рецесивний дефіцит GTP-циклогідроксилази і PTPS зумовлені порушенням біосинтезу ВН4, унаслідок чого виникає гіперфенілаланінемія. За дефіциту сепіаптеринредуктази виникнення гіперфенілаланінемії нехарактерне [9]. Дефіцит DHPR і PCD, зумовлений порушенням регенерації ВН4, також спричиняє виникнення гіперфенілаланінемії. За наявності автосомно-рецесивного GTPCH-дефіцитного синдрому гіперфеніл–аланінемії може не бути [10]; за цієї патології описані кілька клінічних спостережень дофамінзалежної дистонії.
Виділяють 4 з 6 нозологічних форм ВН4-дефіцитних синдромів, при яких розвивається гіперфенілаланінемія (табл. 1) [11]. Клінічні, біохімічні та молекулярно-генетичні дані клінічних спостережень взяті в базах BIODEF, інформація доступна на сайті: www.bh4.org (http://www.biopku.org/home/biodef.asp).
За даними бази BIOMED (1023 клінічних спостереження ВН4-дефіцитних синдромів різного типу), PTPS виявлений у 65,3 % пацієнтів, DHPR — у 24,9 %, GTPCH — у 3,3 %, PCD — у 2,6 % [11, 12]. ВН4-дефіцитні синдроми, що є рідкісними вродженими порушеннями метаболізму, спричиненими недостатністю нейротрансмітерів, за відсутності своєчасного лікування призводять до прогресуючої, нерідко летальної, енцефалопатії.
Основні клінічні прояви ВН4-дефіцитних синдромів: затримка розумового розвитку, судоми, прогресування екстрапірамідних симптомів, порушення тонусу м’язів і пози, ковтання, гіперсалівація — частіше за все виникають протягом перших місяців життя дитини [13].
Оскільки ВН4 — кофактор фенілаланінгідроксилази, для більшості клінічних спостережень за цієї патології характерне підвищення рівня фенілаланіну або гіперфенілаланінемія. Фенілкетонурія — класичне, найбільш відоме захворювання, що супроводжується гіперфенілаланінемією. Таким чином, гіперфенілаланінемії — велика гетерогенна група захворювань, до якої, крім фенілкетонурії, згідно із сучасною класифікацією, включені і ВН4-дефіцитні синдроми [14].
Упровадження в медичну практику ранньої диференціальної клінічної та лабораторної діагностики гіперфенілаланінемій дозволить запобігти розвитку летальних енцефалопатій та суттєво підвищити ефективність лікування цих нейрометаболічних захворювань.
Матеріали та методи
Під час даного дослідження було обстежено 59 пацієнтів із діагнозом PKU віком від 2 до 16 років. Гіперфенілаланінемія була загальним симптомом і встановлена в усіх обстежених нами пацієнтів. Усім хворим було проведено молекулярно-генетичне обстеження на панель мутацій в гені РАН (R408W, Y414C (12-й екзон), R158Q (5-й екзон), Ivs10nt546 (5-й інтрон), Ivs12nt1 (12-й інтрон), R252W, R261W, R261Q, R261X, G272X, P281L, S273F (7-й екзон)). Аналіз проводився в Центрі молекулярної діагностики МОЗ України. Також усі хворі були обстежені на спектр птеринів (біоптерин та неоптерин) в крові та сечі за методикою N. Blau [14]. Пацієнти були розподілені на дві групи: 1-ша група — 42 хворі з діагнозом «фенілкетонурія», у яких у гені фенілаланінгідроксилази виявлені мажорні мутації; дієтотерапія забезпечувала добрий клінічний ефект; 2-га група — 17 хворих із гіперфенілаланінемією, у яких також діагностована фенілкетонурія, але, за даними досліджень ДНК, мутації в гені фенілаланінгідроксилази не виявлені; клінічна картина захворювання у пацієнтів 2-ї групи різнилася. У 4 хворих дієтотерапія виявилася неефективною, незважаючи на нормалізацію рівня фенілаланіну; також відзначали прогресування неврологічних розладів, зокрема порушень тонусу м’язів і пози, гіперкінетичний синдром. У цих 4 пацієнтів додатково визначали активність дигідроптеринредуктази у сухих краплях крові (дослідження проведене в університетській клініці, м. Гейдельберг, Німеччина). Двом пацієнтам проводилося вимірювання концентрації гомованілової кислоти та 5-гідроксііндолоцтової кислоти в спинномозковій рідині методом високоефективної рідинної хроматографії на приладі Dionex Ultimate 3000 з електрохімічною детекцією на детекторі Dionex Coulochem III за протоколом, запропонованим Simon J.R. Heales [15].
Усім пацієнтам призначена дієтотерапія з використанням продуктів спеціалізованого лікувального харчування, амінокислотними сумішами, які не вміщують Phe.
Результати
Аналіз клінічних даних та результатів лабораторних обстежень показав, що пацієнти можуть бути розподілені на дві групи. Перша група — 42 хворі з діагнозом «фенілкетонурія», у яких в гені РАН виявлені мутації; дієтотерапія забезпечувала добрий клінічний ефект. Дослідження рівня птеринів у крові та сечі в пацієнтів із першої групи патологічних відхилень не виявило. Таким чином, причина розвитку гіперфеніл–аланінемії в пацієнтів першої групи була обумовлена мутаціями в гені PAH, діагноз «фенілкетонурія» в цих пацієнтів був беззаперечний.
Друга група — 17 хворих із гіперфенілаланінемією, у яких також діагностовано фенілкетонурію, але за результатами молекулярно-генетичних досліджень мутації в гені PAH виявлені не були; клінічна картина захворювання у пацієнтів 2-ї групи різнилася. Так, у чотирьої хворих дієтотерапія була неефективною. Незважаючи на нормалізацію рівня Phe та своєчасно розпочате лікування, у пацієнтів розвинулися тяжкі неврологічні порушення, зокрема порушення м’язового тонусу та постави, гіперкінетичний синдром. Серед пацієнтів другої групи у 3 осіб виявлені зміни вмісту птеринів як у крові, так і в сечі. Крім того, ще один пацієнт другої групи спостереження демонстрував прогресуючу неврологічну симптоматику та мав обтяжений клініко-генеалогічний анамнез. Виявлені чотири пацієнти були додатково обстежені на активність DHPR. Таким чином, у чотирьох пацієнтів другої групи спостереження були верифіковані діагнози гіперфенілаланінемії, обумовленої ВН4-дефіцитними синдромами, а діагноз фенілкетонурії було знято. У трьох пациєнтів було встановлено діагноз «синдром дефіциту PTPS», а в одного — «синдром дефіциту DHPR». Двом пацієнтам із встановленим діагнозом ВН4-дефіцитних синдромів було проведене дослідження спинномозкової рідини на кількісне визначення гомованілової (HV) та 5-гідроксііндолоцтової кислот (HIIV) з метою корекції лікування.
Клінічне спостереження 1
Пацієнтка Т.Ю., 12 років, діагноз «фенілкетонурія, атипова, злоякісна форма». На момент звернення до Центру орфанних захворювань батьки пацієнтки скаржилися на неефективність дієтотерапії, грубу затримку психомоторного розвитку дитини, судомний синдром на тлі різко зниженого тонусу м’язів, слинотечу.
Анамнез: дитина народилася від І патологічної вагітності (анемія, загроза переривання у І триместрі), нормальних фізіологічних пологів на 38–39-му тижні гестації, маса тіла при народженні 2400 г. До 2 міс. дитина намагалася піднімати голову, однак була гіпотонічною, що пояснювали фізіологічною незрілістю. У віці 2 місяців виник судомний синдром, що не вдавалося ефективно лікувати. У віці 9 міс. у пацієнтки виявлена гіперфенілаланінемія — рівень фенілаланіну 12 мг% (норма — не більше 2 мг%); діагностована фенілкетонурія. Призначена дієтотерапія — спеціальні амінокислотні суміші, які не містять фенілаланіну. Нормалізація рівня фенілаланіну не сприяла покращенню неврологічного статусу: зберігалися виражена затримка психомоторного розвитку, порушення тонусу м’язів, епісиндром, гіперсалівація. Проведене дослідження біоптеринового профілю в крові і сечі, активності дигідроптеринредуктази. Рівен біоптерину був різко знижений — 0,02 nmol/gHb (норма — 0,5–3 nmol/gHb); рівень неоптерину становив 2,45 nmol/gHb (норма — 0,3–4 nmol/gHb), уміст біоптерину В/(N + В) × 100 — різко зменшений — 12 % (норма — 44–77 %); активність DHPR — 2,8 mU/mg Hb (норма). Установлений діагноз «ВН4-дефіцитний синдром: дефіцит 6-пірувол-тетрагідробіоптеринсинтази (PTPS-синдром)».
Призначений сапроптерину дигідрохлорид у дозі 100 мг/добу, L-dopa/carbidopa, 5-гідрокситриптофан. Через 3 міс. судомні напади припинилися, практично зникла слинотеча, дитина добре утримує голову, зберігає позу в положенні сидячи. Зберігається виражена затримка психічного й мовленнєвого розвитку, що, ймовірно, пояснюється пізнім початком лікування. Спеціальна дієта не рекомендована, рівень фенілаланіну в нормі.
Клінічне спостереження 2
Пацієнт Б.А., 3 роки 2 міс., направлений на консультацію до Центру орфанних захворювань з діагнозом «дегенеративне захворювання ЦНС при фенілкетонурії» через неефективність лікування за нормального рівня фенілаланіну.
Скарги батьків на відсутність у дитини цікавості до іграшок, самостійної ходьби, контролю пози в положенні сидячи, виражене зниження м’язового тонусу, слинотечу.
Анамнез: дитина народилася від І нормальних вагітності і пологів, маса тіла при народженні — 3530 г. Гіперфенілаланінемія виявлена під час масового неонатального скринінгу, рівень фенілаланіну 10 мг% (норма — не більше 2 мг%). Дієтотерапія розпочата у віці 4 тижнів із використанням спеціалізованих лікувальних сумішей, що не містять фенілаланіну. На дієтотерапії рівень фенілаланіну нормалізувався та не перевищував рекомендованих концентрацій — 2–4 мг%. Проте, незважаючи на добрий контроль рівня фенілаланіну та своєчасно розпочату дієтотерапію, у пацієнта спостерігались виражена затримка психомоторного розвитку, зниження тонусу м’язів, рухові порушення за типом хореоподібних гіперкінезів, гіперсалівація. Крім того, у віці 1 рік 4 міс. у дитини виникли поліморфні судоми за типом генералізованих складних абсансів і міоклоній (напади до 5–10 разів на добу). Протисудомна терапія була неефективна. Таким чином, клінічна картина захворювання мала прогредієнтний перебіг.
Проведений ДНК-аналіз на панель мутацій у гені — мутації не виявлені.
Птериновий профіль у сечі: рівень біоптерину різко знижений — 0,04 nmol/gHb (норма — 0,5–3 nmol/gHb); рівень неоптерину становив 1,45 nmol/gHb (норма — 0,3–4 nmol/gHb); уміст біоптерину В/(N + В) × 100 — різко зменшений — 2 % (норма — 44–77 %). Активність DHPR у нормі — 2,8mU/mg Hb. Установлений діагноз «ВН4-дефіцитний синдром: дефіцит 6-пірувол-тетрагідробіоптеринсинтази (PTPS-синдром)».
Рекомендоване специфічне лікування прекурсорами біогенних нейротрансмітерів L-dopa (+10% carbidopa) у дозі 2–3 мг/кг, яку поступово підвищували до 7–8 мг/кг; 5-гідрокситриптофан у дозі 2–3 мг/кг/добу, яку підвищували до 5–6 мг/кг/добу. На жаль, через неможливість лікування синтетичним ВН4 (сапроптерину дигідрохлоридом) дитина перебуває на рестрикційній дієті, що негативно позначається на її фізичних даних: дефіцит маси тіла пацієнта 1,8 σ відхилення. Через 6 міс. від початку лікування напади судом припинилися, дитина утримує голову, здатна зберігати позу в положенні сидячи, намагається самостійно ходити.
З метою корекції лікування проведена діагностична спинномозкова пункція, для визначення концентрації метаболітів нейротрансмітерів: гомованілова кислота (HVA) — 85,7 нмоль/л (норма — 154–867 нмоль/л), 5-гідроксііндолоцтова кислота (5HIAA) — 23 нмоль/л (норма 89–367 нмоль/л). Таким чином, було виявлено, що доза прекурсорів біогенних амінів потребує корекції й подальшого поступового збільшення до 8–10 мг/кг.
У наведеному клінічному спостереженні застосування сапроптерину гідрохлориду дозволило б контро–лювати рівень фенілаланіну без дотримання дієти та суттєво розширити вживання природного білка, що сприяло б збільшенню м’язової маси та покращенню фізичних даних пацієнта.
Клінічне спостереження 3
Пацієнт Ш.Д., 2 роки 1 міс., направлений на консультацію у віці 6 місяців до Центру орфанних захворювань з діагнозом «фенілкетонурія» через неефективність лікування: виражена затримка психомоторного розвитку, синдром рухових порушень, епілептичний синдром за нормального рівня фенілаланіну.
Скарги батьків на відсутність у дитини цікавості до оточуючих предметів, затримку психомоторного розвитку (не утримує голову, не перевертається), виражені рухові порушення у вигляді тонічного повороту голови, вигинання тіла, підвищення м’язового тонусу, порушення рухів очних яблук у вигляді підкатувань.
Анамнез: дитина народилася від ІІ нормальних вагітності і пологів, маса тіла при народженні — 2810 г, довжина — 50 см; шкала Апгар — 8/8 балів. Перша вагітність закінчилась самовільним викиднем у терміні 7–9 тижнів. Гіперфенілаланінемія виявлена під час масового неонатального скринінгу, рівень феніл–аланіну 14 мг% (норма — не більше 2 мг%). Дієтотерапія розпочата у віці 3 тижнів — спеціалізовані суміші, що не містять фенілаланіну. На дієтотерапії рівень фенілаланіну не перевищує 2–4 мг%, що є показником ефективного розрахунку та дотримання батьками дитини рекомендацій дієтолога. Дитина добре набирала вагу, проте, незважаючи на добрий контроль рівня фенілаланіну, у пацієнта спостерігалася виражена затримка психомоторного розвитку, порушення м’язового тонусу за типом спастичного тетерапарезу, на тлі якого відмічалися гіперкінетичний синдром, гіперсалівація, тонічні гіперкінези очних яблук, які мама дитини помітила з перших тижнів життя.
Проведений ДНК-аналіз на панель мутацій у гені фенілаланінгідроксилази — патології не виявлено.
Птериновий профіль у сечі: рівень біоптерину різко знижений — 0,23 nmol/gHb (норма — 0,3–4 nmol/gHb); рівень неоптерину становив 2,43 nmol/gHb (норма — 0,3–4 nmol/gHb); уміст біоптерину В/(N + В) × 100 — різко зменшений — 8,6 % (норма — 44–77 %). Активність DHPR у нормі — 2,4 mU/mg Hb.
Встановлений діагноз «ВН4-дефіцитний синдром: дефіцит 6-пірувол-тетрагідробіоптеринсинтази (PTPS-синдром)».
Рекомендоване специфічне лікування прекурсорами біогенних нейротрансмітерів L-dopa (+10% carbidopa) в дозі 1–3 мг/кг, яку поступово підвищували до 5–6 мг/кг; 5-гідрокситриптофан у дозі 2–3 мг/кг/добу, яку підвищували до 5–6 мг/кг/ добу. Сапроптерину дигідрохлорид отримує з 6,5 місяця, що дозволяє без дієтотерапії утримувати рівень Phe в нормальних значеннях. Через 3 міс. від початку лікування дитина почала утримувати голову, з’явилась цікавість до іграшок, у віці 1 рік стала здатна зберігати позу в положенні сидячи, почала самостійно перевертатись, припинились тонічні гіперкінези очних яблук та вигинання тулуба, почала брати в руки предмети. Але затримка психомоторного розвитку зберігається, відмічаються гіперкінези язика, відсутній активний словниковий запас.
Із метою корекції лікування проведена діагностична спинномозкова пункція для визначення концентрації метаболітів нейротрансмітерів: гомованілова кислота (HVA) — 45,63 нмоль/л (норма — 154–867 нмоль/л), 5-гідроксііндолоцтова кислота (5HIAA) — 0 (норма — 89–367 нмоль/л). Таким чином, було виявлено, що доза прекурсорів біогенних амінів потребує корекції й подальшого поступового збільшення до 8–10 мг/кг.
Клінічне спостереження 4
Пацієнт М.Д., 2 років. На момент звернення до Центру орфанних захворювань батьки хворого скаржилися на втрату набутих навичок: дитина перестала утримувати голову, сидіти, стояти, втратила цікавість до іграшок, крім того, у неї виникли підкатування очних яблук і часті напади тонічних судом, резистентних до протисудомної терапії. Клініко-генеалогічний анамнез обтяжений: старший брат хворого помер у віці 5 років 8 міс., діагноз «лейкодистрофія, епісиндром».
Дитина народилася від ІІ нормальної вагітності, фізіологічних пологів. Маса тіла при народженні — 3200 г, оцінка за шкалою Апгар 8–9 балів. За даними масового неонатального скринінгу виявлений підвищений рівень фенілаланіну — 16 мг% (норма — не більше 2 мг%). Діагноз «фенілкетонурія» встановлений у віці 2 тижнів. Дієтотерапія розпочата у віці 3 тижнів, що дозволило нормалізувати і протягом першого року життя пацієнта підтримувати рекомендований рівень фенілаланіну — 1,7–2,5 мг%. Упродовж першого року життя розвиток дитини характеризувався незначною затримкою статомоторного розвитку: у 4 міс. вона утримувала голову, у 9 міс. — самостійно сіла, в 1 рік і 2 міс. почала ходити з підтримкою.
У віці 1 рік 2 міс. у дитини виник епісиндром, на тлі якого впродовж 10 міс. всі набуті навички були втрачені (рис. 1).
За даними молекулярно-генетичного аналізу на панель мутацій в гені РАН патологічних мутацій не виявлено. Птериновий профіль в крові: неоптерин — 0,23 nmol/gHb (норма — 0,19–2,93 nmol/gHb); біоптерин — 0,28 nmol/gHb (норма — 0,08–1,2 nmol/gHb); уміст біоптерину В = В/(N + В) × 100 — 55 % (норма — 16,0–64,7 %). Птериновий профіль у сечі в нормі. Активність DHPR у сухій краплі крові — 0, відсутня (норма — понад 1,1 mU/mgHb).
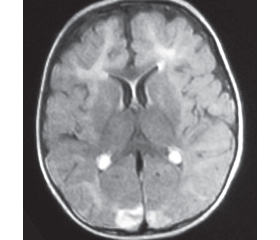
За даними магнітно-резонансної томографії головного мозку виявлені його атрофічні зміни, порушення структури білої речовини (рис. 2).
/123-2.jpg)
Таким чином, з урахуванням різкого зниження активності DHPR, наявності гіперфенілаланінемії, характерної клінічної картини, прогредієнтного перебігу захворювання встановлений діагноз «ВН4-дефіцитний синдром: дефіцит дигідроптеринредуктази (DPHR-синдром)». Рекомендоване лікування прекурсорами нейротрансмітерів: L-dopa (+10% carbidopa) в дозі 2–3 мг/кг, із поступовим її підвищенням до 7–8 мг/кг; 5-гідрокситриптофан у дозі 2–3 мг/кг/добу, з поступовим підвищенням до 5–6 мг/кг/добу; фолінова кислота в дозі 15–20 мг/добу; дієтотерапія — вживання продуктів спеціального лікувального харчування, що не містять фенілаланіну. На тлі зазначеного лікування стан дитини покращився: практично припинилися дистонічні атаки, хлопчик став спокійнішим, але через пізній початок лікування суттєвого відновлення втрачених статомоторних, психічних і мовних навичок досягти не вдалося.
Дані літератури щодо ефективності сапроптерину дигідрохлориду при DHPR-синдромі різняться; незаперечним є застосування дієтотерапії в лікуванні таких пацієнтів, що зумовлене вираженим пошкоджуючим впливом фенілаланіну на мозок хворого.
Обговорення
За даними літератури, ВН4-дефіцитні синдроми характеризуються тяжкими неврологічними розладами, типовими для дефіциту катехоламінів і серотоніну [16]. Зокрема, дефіцит дофаміну клінічно проявляється гіподинамією, паркінсонізмом, дистонією, гіперсалівацією, утрудненням ковтання, страбізмом, тонічним гіперкінезом очних яблук. Дефіцит серотоніну характеризується депресією, порушенням терморегуляції, інсомнією; дефіцит норадреналіну — аксіальною гіпотонією, птозом, мозочковими та іншими неврологічними симптомами.
Виникнення неврологічних симптомів при ВН4-дефіцитних станах зумовлене дефіцитом дофаміну, норадреналіну і серотоніну, проте ці симптоми (затримка розумового розвитку, судоми, зниження тонусу м’язів, дистонія та інші ексрапірамідні симптоми) неспецифічні, вони характерні для багатьох неврологічних захворювань [17]. Пацієнтів направляли до Центру орфанних захворювань з попереднім діагнозом «дегенеративне захворювання ЦНС», який не пов’язували з фенілкетонурією і тим більше з гіперфенілаланінеміями (гетерогенною групою захворювань, у діагностиці яких необхідний диференційний підхід). В одного з трьох пацієнтів встановлений попередній діагноз «злоякісна форма фенілкетонурії», проте це не відповідає сучасній класифікації [9, 14].
Дефіцит 6-пірувол-тетрагідробіоптеринсинтази (PTPS-синдром) і дигідроптеринредуктази (DHPR-синдром) — найбільш поширені форми гіперфеніл–аланінемій [12]. Саме ці синдроми діагностовані нами в пацієнтів при гіперфенілаланінемії.
PTPS-синдром майже у 80 % клінічних спостережень має тяжкий перебіг, лише у небагатьох хворих відзначають м’який, периферичний фенотип [17].
Троє обстежених нами хворих при PTPS-синдромі народилися в належні строки, однак маса тіла при народженні одного з них була нормальною, а двох — низькою. За даними літератури, недоношеність і/або низька маса тіла при народженні характерні для хворих при PTPS-синдромі [18].
Дуже важливо відзначити помірно підвищений рівень фенілаланіну у крові пацієнтів при ВН4-синдромах: за даними масового неонатального скринінгу, він не перевищує 15 мг%, за класичної фенілкетонурії рівень фенілаланіну перевищує 20 мг% [19].
Птериновий профіль у сечі пацієнтів при PTPS-синдромі такий: характерне зниження рівня неоптерину та біоптерину. Біоптериновий профіль у сечі хворих при DHPR-синдромі в нормі, тому обов’язковим є визначення активності дигідроптеринредуктази в крові пацієнтів при гіперфенілаланінемії [20].
В усіх обстежених нами пацієнтів дієтотерапія виявилася неефективною, відзначали прогресування неврологічних симптомів на тлі контрольованого рівня фенілаланіну у крові. Клініко-генеалогічний анамнез хворого при DHPR-синдромі був обтяжений: старший брат пацієнта помер від невідомого прогресуючого захворювання ЦНС із гіперфенілаланінемією. Таким чином, ВН4-дефіцитні синдроми — захворювання, які загрожують життю, особливо якщо лікування розпочате пізно [13, 21].
Застосування прекурсорів нейротрансмітерів (L-dopa, 5-гідрокситриптофан), сапроптерину дигідрохлориду сприяло суттєвому покращенню стану пацієнтів. На жаль, через несвоєчасну діагностику і, відповідно, пізній початок лікування ефективність терапії виявилася нижчою порівняно з такою за даними літератури [22].
Висновки
Гіперфенілаланінемія — основний прояв гетерогенної групи рідкісних нейрометаболічних захворювань, які вимагають подальшої діагностичної верифікації. У новонароджених, у яких під час проведення масового неонатального скринінгу на фенілкетонурію виявлений підвищений рівень фенілаланіну, необхідно застосовувати додатковий діагностичний протокол (визначення птеринового профілю в крові та сечі, обов’язкове дослідження активності дигідроптеринредуктази у сухій краплі крові).
Дослідження птеринів необхідне в пацієнтів із раніше встановленим діагнозом «фенілкетонурія» за неефективності дієтотерапії, доброго контролю рівня фенілаланіну, відсутності позитивної клінічної динаміки, прогресування когнітивних і неврологічних порушень, зниження тонусу м’язів, виникнення судомного синдрому, наявності екстрапірамідних симптомів.
На сьогодні ВН4-дефіцитні синдроми курабельні. Рання діагностика та своєчасно розпочате лікування дозволяють суттєво покращити стан хворих, а в деяких ситуаціях досягти практично повного відновлення втрачених функцій.
Для деяких ВН4-дефіцитних синдромів гіперфенілаланінемія не характерна, що унеможливлює раннє виявлення таких пацієнтів під час неонатального скринінгу. Тому виявлення прогресуючих неврологічних симптомів у пацієнтів віком до 1 року й у ранньому дитинстві, які супроводжуються втратою когнітивних функцій на тлі м’язової гіпотонії, дистонічних порушень і судом, зумовлює необхідність дослідження біоптеринового профілю в крові і сечі.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
Список литературы
1. Blau N. Hyperphenylalaninemias: disorders of tetrahydrobiopterin metabolism / N. Blau, B. Thöny // Pediatric endocrinology and inborn errors of metabolism / K. Sarafoglou, G.F. Hoffmann, K.S. Roth editors. — New York: McGraw-Hill Med., 2009. — P. 170-175.
2. Hyperphenylalaninemia due to a deficiency of biopterin. A variant form of phenylketonuria / S. Kaufman, S. Berlow, G.K. Summer, S. Milstien, J.D. Schulman, S. Orloff, S. Spielberg, S. Pueschel // N. Engl. J. Med. — 1978. — V. 299(13). — P. 673-679.
3. Tetrahydrobiopterin therapy of atypical phenylketonuria due to defective dihydrobiopterin biosynthesis / J. Schaub, S. Däumling, H.C. Curtius, A. Niederwieser, K. Bartholomé, M. Viscontini, B. Schircks, J.H. Bieri // Arch. Dis. Child. — 1978. — V. 53(8). — P. 674-676.
4. Disorders of tetrahydrobiopterin and related biogenic amines / N. Blau, B. Thöny, R.G.H. Cotton, K. Hyland // The metabolic and molecular bases of inherited disease / C.R. Scriver, A.L. Beaudet, W.S. Sly, D. Valle editors. — N.Y.: McGraw-Hill Med., 2001. — P. 1725-1776.
5. Werner E.R. Tetrahydrobiopterin: biochemistry and pathophysiology / E.R. Werner, N. Blau, B.Thöny // Biochem. J. — 2011. — Vol. 438. — P. 397-414.
6. Longo N. Disorders of BH4 metabolism / N. Longo // J. Inherit. Metab. Dis. — 2009. — Vol. 32. — P. 333-342.
7. Thöny B., Blau N., Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterine synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteredine reductase genes / B. Thöny, N. Blau // Hum. Mutat. — 2006. — Vol. 27. — P. 870-878.
8. Mutations in the sepiapterin reductase reductase gene cause a novel tetrahydrobiopterin-dependent monoamine neurotransmitter deficiency without hyperphenylalaninemia / L. Bonafé, B. Thöny, J.M. Penzien, B. Czarnecki, N. Blau // Am. J. Hum. Genet. — 2001. — Vol. 69. — P. 269-277.
9. Clinical and biochemical characterization of patient with early infantile onset of autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia / T. Opladen, G. Hoffmann, F. Hörster, A.B. Hinz, K. Neidhardt, C. Klein, N. Wolf // Mov. Disord. — 2011. — Vol. 26(1). — P. 157-161.
10. Porta F., Mussa A., Concolino D., Spada M., Ponzone A. Dopamine agonists in 6-pyruvoyl tetrahydropterin synthase deficiency // Neurology. — 2009. — Vol. 73. — P. 633-637.
11. BIODEF-International Database of Tetrahydrobiopterin Deficiencies [Internet] // Available at: biopku.org: Databases of Pediatric Neurotransmitter Disorders inc. PKU www.biopku.org accessed May 2010.
12. Opladen T. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia / T. Opladen, G.F. Hoffmann, N. Blau // J. Inherit. Metab. Dis. — 2012. — V. 35, 6. — P. 963-973.
13. Phenylketonuria and BH4 deficiencies / N. Blau, B.K. Burton, B. Thöny, F. J. van Spronsen, S. Waisbren. — 1st еd. — Bremen: UNI-MED, 2010. — 80 p.
14. Burlina A., Blau N. Tetrahydrobiopterin disorders presenting with hyperphenylalaninemia / Burlina A., Blau N. // Congenital neurotransmitter disorders. A clinical approach / G.F. Hoffmann, N. Blau editors. — N.Y.: Nova Biomedical, 2014. — P. 39-50.
15. Heales Simon J.R. Laboratory guide to the methods in biochemical genetics / B. Thöny, M. Duran, K.M. Gibson editors. — Berlin; Heidelberg: Springer-Verlag, 2008. — Р. 703-715.
16. Van Spronsen F.J. Brain dysfunction in phenylketonuria: is phenylalanine toxicity the only possible cause? / F.J. van Spronsen, M. Hoeksma, D.J. Reijngoud // J. Inherit. Metab. Dis. — 2009. — V. 32, 1. — P. 46-51.
17. Reblova K., Hruba Z., Prochazkova D., Pazdirkova R., Pouchla S., Fajkusova L. Hyperphenylalaninemia in the Czech Republic: genotype-phenotype correlations and in silico analysis of novel missense mutations // Clin. Chim. Acta. — 2013. — V. 419. — P. 1-10.
18. Phenotypic variability, neurological outcome and genetics background of with 6-pyruvoyl-tetrahydropterine synthase deficiency / V. Leuzzi, C.A. Carducci, C.L. Carducci, S. Pozzessere, A. Burlina, R. Cerone, D. Concolino, M.A. Donati, L. Fiori, C. Meli, A. Ponzone, F. Porta, P. Strisciuglio, I. Antonozzi, N. Blau // Clin. Genet. — 2010. — V. 77. — P. 249-257.
19. Blau N. Phenylketonuria / N. Blau, F.J. van Spronsen, H.L. Levy // Lancet. — 2010. — V. 376, 9750. — P. 1417-1427.
20. Diagnosis of tetrahydrobiopterin deficiency using filter paper blood spots: further development of method and 5 years experience / T. Opladen, B. Abu Seda, A. Rassi, B. Thöny, G.F. Hoffmann, N. Blau // J. Inherit. Metab. Dis. — 2011. — V. 34(3). — P. 819-826.
21. Blau N. Genetics of phenylketonuria: then and now / N. Blau // Hum. Mut. — 2016. — Vol. 37(6). — P. 508-515.
22. Jaggi L. et al. Outcome and long-term follow-up of 36 patients with tetrahydrobiopterin deficiency // Mol. Gen. Metab. — 2008. — Vol. 93. — P. 295-305.

/120-1.jpg)
/123-1.jpg)