
Международный неврологический журнал 6 (68) 2014
Вернуться к номеру
Биопрепараты и прионные болезни: возможна ли этиологическая связь?
Авторы: Бойченко М.Н., Волчкова Е.В., Пак С.Г. - ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова», Минздравсоцразвития России, г. Москва
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
В обзоре литературы отражены современные данные зарубежных исследователей о природе и биологических свойствах прионных белков, об их конформационных разновидностях. Приведены сведения о роли клеточного прионного белка в функционировании различных тканей и систем организма, путях инфицирования аномальными изоформами прионов. Описаны патоморфологические изменения и клинические проявления прионных заболеваний человека, дана их современная классификация. Подробно описаны способы профилактики и предупреждения распространения прионных болезней, а также современные методы лабораторной диагностики. Дана оценка вероятности заражения в результате применения того или иного биопрепарата в зависимости от характеристики входящих в его состав агентов и метода обработки нативного материала, послужившего основой для его создания.
В огляді літератури відображені сучасні дані зарубіжних дослідників про природу і біологічні властивості пріонних білків, про їх конформаційні різновиди. Наведено відомості про роль клітинного пріонного білка у функціонуванні різних тканин і систем організму, шляхи інфікування аномальними ізоформами пріонів. Описано патоморфологічні зміни і клінічні прояви пріонних захворювань людини, дана їх сучасна класифікація. Докладно описані способи профілактики та запобігання поширенню пріонних хвороб, а також сучасні методи лабораторної діагностики. Дано оцінку ймовірності зараження в результаті застосування того чи іншого біопрепарату залежно від характеристики агентів, що входять до його складу, і методу обробки нативного матеріалу, що послужив основою для його створення.
The literature review reflects the current data of foreign researchers on the nature and biological properties of the prion proteins, their conformational variations. Information about the role of cellular prion protein in the functioning of various tissues and body systems, and ways of infection by abnormal prion isoforms is presented. Pathological changes and clinical manifestations of prion diseases in humans are described; current classification of prion diseases is presented. Measures for prophylaxis and prevention of prion diseases’ extension as well as modern methods of laboratory diagnosis are described in detail. The probability of infection through the use of a biological preparation, depending on the characteristics of its constituent agents and the processing method of the native material, which served as the basis for its creation, is estimated.
прионы, клеточные прионные белки, изоформы прионного белка, механизм образования конформационных изоформ прионного белка, прионные заболевания, биопрепараты, лабораторная диагностика.
пріони, клітинні пріонні білки, ізоформи пріонів білка, механізм утворення конформаційних ізоформ пріонів білка, пріонні захворювання, біопрепарати, лабораторна діагностика.
cellular prion proteins, prion protein isoforms, mechanism of conformational conversion of the prion protein into isoform, prion diseases, biological preparations, laboratory diagnostics.
Статья опубликована на с. 77-84
В последние годы отмечен рост числа биопрепаратов, для изготовления которых все шире используются различные ткани животных, в том числе коров и овец, то есть животных, потенциально являющихся источником целой группы прионных заболеваний. В связи с этим актуальным становится вопрос обеспечения и оценки безопасности этих препаратов для человека. Чтобы разобраться в столь сложном вопросе, необходимо проанализировать имеющиеся сведения об этиологии, эпидемиологии, патогенезе этой группы заболеваний, накопленные за последние десятилетия. Большие успехи, достигнутые за последние 10–15 лет в области изучения прионов и вызываемых ими заболеваний, обосновали естественную потребность в систематизации накопленных данных. В табл. 1 представлена современная классификация прионных болезней человека и животных.
/78/78.jpg)
История прионных заболеваний берет свое начало в области ветеринарии. В 1933 г. для развития каракулеводства исландские фермеры закупили в Германии большую партию овец. Через несколько лет среди закупленных животных стали регистрироваться случаи заболевания, названного «скрепи овец» (от англ. scrappy — лоскутный), принявшего массовый характер и имевшего быстрый летальный исход. Причину этого заболевания впервые изучил доктор B. Sigurdsson. Он сформулировал четыре основных признака, позволивших ему ввести новый термин для обозначения этой группы заболеваний — «медленные инфекции», и в 1954 г. впервые прочитал цикл лекций в Лондонском университете [1].
Эти признаки и легли в основу характеристики медленных инфекций:
— продолжительный инкубационный период;
— медленный прогрессирующий характер течения;
— необычность поражения органов и тканей;
— неизбежность смертельного исхода.
Через 3 года американский ученый D.C. Gajdusek описывает заболевание, которое встречается в горных районах острова Новая Гвинея среди папуасов-каннибалов и сейчас известно под названием «куру» [2]. Эта болезнь характеризовалась многолетним (до 30 лет) инкубационным периодом, медленным прогрессирующим течением, поражением только головного мозга и смертельным исходом. Таким образом, проблема медленных инфекций из ветеринарии перетекла в область медицины.
Картина поражений при медленных инфекциях отличается своеобразием. Патологические изменения, обнаруживаемые в центральной нервной системе (ЦНС), характеризуются тем, что в головном, а иногда и в спинном мозге в отсутствие воспаления наблюдается гибель нервных клеток и их отростков. В тканях мозга появляются вакуоли с постепенным развитием «губкообразного» состояния. В мозговой ткани также накапливаются амилоидные бляшки, разрастается глиозная ткань. Воспаление и иммунный ответ отсутствуют. Кроме куру к середине 1980-х гг. стало известно еще о нескольких подобных заболеваниях человека (болезнь Крейтцвельда — Якоба, синдром Герстмана — Штраусслера — Шейнкера, смертельная семейная бессонница), а также животных (трансмиссивная энцефалопатия норок, хроническая изнуряющая болезнь оленей и лосей). Подобное своеобразие патоморфологических изменений в нервной ткани определило название этой группы — «трансмиссивные губкообразные энцефалопатии». Было установлено, что возбудитель не размножается на искусственных питательных средах, проходит через бактериальные фильтры, не виден в световой микроскоп, но в то же время устойчив к ультрафиолетовому излучению, кипячению в течение 15 минут и нуклеазам. То есть его нельзя было отнести ни к вирусам, ни к бактериям, ни к вироидам. Этиология была установлена американским ученым S.B. Prusiner, который показал, что трансмиссивные губкообразные энцефалопатии связаны с инфицированием низкомолекулярным белком, не содержащим никаких нуклеиновых кислот. Обнаруженный белок был назван прионом [3]. За открытие прионов в 1997 г. S.B. Prusiner был удостоен Нобелевской премии.
В настоящее время принято считать, что прионы — это белковые инфекционные частицы, возбудители прионных конформационных болезней, которые развиваются в результате неправильного сворачивания (нарушения правильной конформации) клеточного белка, необходимого для нормального функционирования организма. Название произошло от английского словосочетания proteinaceous infectious particles — белковые инфекционные частицы.
Следует особо отметить, что прионовый протеин PrPС (cellularprion protein) — нормальная изоформа прионного белка с молекулярной массой 33–35 кД, детерминируемая геном прионного белка (PrNP), расположенного на 20-й хромосоме человека, по своей природе является сиалогликопротеином и синтезируется главным образом в нейронах, хотя продуцировать его могут и многие другие клетки [4, 5], причем этот нормальный PrPc локализован на поверхности клетки. Он как бы «заякорен» в богатую холестеролом мембрану клетки через молекулу гликопротеина [6], и его отличительной особенностью является чувствительность к протеазе. PrPc поддерживает качество миелиновой оболочки, регулирует передачу нервных импульсов, циркадные (суточные) ритмы, процессы окисления, участвует в метаболизме меди в ЦНС и в регуляции деления стволовых клеток костного мозга. PrPc обнаружен также в селезенке, лимфатических узлах, коже, желудочно-кишечном тракте и фолликулярных дендритных клетках, роговице глаза. Функции прион-протеина в организме здорового человека до конца не изучены до сих пор. Помимо вышесказанного известно, что он участвует в эндоцитозе и катаболизме клеток. Прион-протеин также необходим для нормальной синаптической функции. Не исключено, что прионы играют определенную роль в межклеточном узнавании и клеточной активации. По мнению некоторых специалистов, нормальные прионы способны подавлять процессы старения, чем и объясняется сходство клинических и морфологических характеристик прионных болезней с геронтологическими заболеваниями ЦНС.
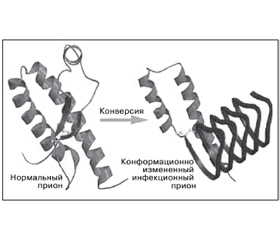
Молекула нормального (клеточного) прионного белка состоит из четырех a-спиральных доменов, стабилизированных междоменными электростатическими взаимодействиями и S-S1-связью (рис. 1) [7, 8].
Измененные модификациями изоформы прионного белка PrPsc (scrapie prion protein — от названия прионной болезни овец скрепи — scrapie) или, например, PrPcjd (при болезни Крейтцфельдта — Якоба) носят патологический характер, способны инфицировать многие виды животных и человека (рис. 1) [9, 10].
/78/78_2.jpg)
Такие измененные прионы устойчивы к протеолизу, излучениям, высокой температуре, формальдегиду, глутаральдегиду, b-пропиолактону [9, 10]. Но самое главное — они способны к агрегации в амилоидные фибриллы, обладающие гидрофобностью [11–13], и формируют нерастворимые агрегаты различных размеров. В результате PrPsc накапливаются в плазматических везикулах клетки.
С учетом структурной близости инфекционного прионного белка и его нормальной (клеточной) изоформы становится понятным, что при развитии прионных заболеваний в организме людей и животных не обнаруживаются антитела на прионный белок PrPsc, который иммунной системой воспринимается как «свой». Это затрудняет лабораторную диагностику заболеваний, их иммунотерапию и иммунопрофилактику [8].
Превращение нормального PrPc в модифицированную изоформу оказывается возможным в результате порой даже незначительных мутационных изменений в PrP-гене, а также под действием самой молекулы инфекционного прионного белка. Превращение PrPc в измененные формы (PrPsc и др.) происходит при нарушении кинетически контролируемого равновесия между ними. Процесс усиливается при возрастании количества патологического прионного белка в самой клетке (PrP) или экзогенного приона. Предполагается, что конверсия индуцируется белком PrPsc, выполняющим роль матрицы. Впервые эта гипотеза была высказана еще в 1967 г. [14] и пересмотрена в 1993-м [15]. Она подтверждается данными, базирующимися на результатах экспериментов, полученных с искусственно синтезированными рекомбинантными прионами в форме b-тяжей [7]. Конформационные изменения связаны с расплетением в С-терминальном участке белка PrPc а-спирали, в результате чего она заменяется на bтяжи [10, 13], и именно этот С-терминальный участок конформационно измененной изоформы PrPsc становится резистентным к протеазе [11–13]. Накопление измененного прионного белка сопровождается его агрегацией, образованием высокоупорядоченных фибрилл (амилоидов), что в конце концов приводит к гибели клетки. Высвободившийся патологически измененный прион, по-видимому, оказывается способным проникать в соседние клетки, также вызывая их гибель. Процесс усиливается при возрастании количества патологического приона, который образует агрегаты с собой и с PrPc на поверхности клетки: в результате PrPc преобразуется в PrPsc, и далее цикл продолжается.
В настоящее время предлагается четыре механизма образования конформационно измененного приона (рис. 2) [16].
Инфицирование аномальными изоформами приона может происходить различными путями:
— при употреблении недостаточно термически обработанных продуктов животного происхождения, например мяса, мозга крупного рогатого скота, больного губкообразной энцефалопатией;
— при трансплантации тканей (например, роговицы глаза, твердой мозговой оболочки), при переливании крови, применении гормонов от лиц, инфицированных патологическими изоформами прионов, а также при введении в организм человека биологически активных веществ животного происхождения; использовании контаминированных или недостаточно простерилизованных хирургических инструментов; при прозекторских манипуляциях;
— через иммунобиопрепараты, не подвергшиеся соответствующей обработке.
Но наиболее частым является пероральное заражение как человека, так и животных. При этом патологические прионы, попав в кишечник, транспортируются в кровь и лимфу. После периферической репликации в селезенке, аппендиксе, миндалинах и других лимфоидных тканях они переносятся в мозг по периферическим нервам (нейроинвазия). Возможно прямое проникновение прионов в мозг — через гематоэнцефалический барьер. Ранее считалось, что ЦНС — это единственная ткань, в которой накапливаются патологические прионы, однако появились исследования, изменившие данную гипотезу. Оказалось, что накопление PrPsc в селезенке связано с увеличением и функционированием фолликулярных дендритных клеток. Прионы, попадая в мозг, накапливаются в нем в больших количествах, вызывая амилоидоз (внеклеточный диспротеиноз, характеризующийся отложением амилоида с развитием атрофии и склероза ткани) и астроцитоз (разрастание астроцитарной нейроглии, гиперпродукция глиальных волокон). В результате формируются фибриллы, агрегаты белка или амилоида и губкообразные изменения мозга (трансмиссивные губкообразные энцефалопатии) [17]. После инфицирования и репликации в ЦНС происходит распространение прионов по периферическим нервам к другим тканям, где происходит вторичная прионная репликация [17]. Секреция прионов из инфицированного организма в окружающую среду происходит с испражнениями, мочой, слюной, грудным молоком, кожными выделениями, что формирует источник прионов в окружающей среде, где они могут сохраняться в неизменном состоянии до 16 лет, создавая стойкие очаги заражения, в частности, на пастбищах, местах содержания животных и т.д. [18, 19].
Клиническая картина прионных болезней человека
Трансмиссивные губкообразные энцефалопатии (ТГЭ) человека в настоящее время хотя и разделяют на три основные группы (инфекционные, спорадические и наследственные), но подобное деление носит весьма приблизительный характер. В группу инфекционных отнесены куру, ятрогенная форма болезни Крейтцфельдта — Якоба (БКЯ, синоним: подострая спонгиоформная энцефалопатия) и так называемый новый вариант БКЯ, впервые описанный в 1996 г. в Англии [20].
В группе спорадических ТГЭ в настоящее время рассматривается только спорадическая БКЯ. И наконец, к наследственным ТГЭ относятся фамильная БКЯ, синдром Герстмана — Штраусслера — Шейнкера и фатальная семейная инсомния.
Инфекционные трансмиссивные губкообразные (спонгиоформные) энцефалопатии
Для болезни куру и ятрогенной формы БКЯ характерны контакт с инфекционным агентом ТГЭ в связи либо с обычаем ритуального поедания мозга умерших на островах Новой Гвинеи (болезнь куру) [2], либо с трансплантацией роговицы или твердой мозговой оболочки или попаданием инфекционного агента с контаминированного нейрохирургического инструмента от пациентов с ТГЭ [21]. Ятрогенная БКЯ была также описана при введении гормона роста, полученного при экстракции гипофиза от большого пула пациентов без установленного диагноза [21]. Первичные клинические проявления при болезни куру и ятрогенной БКЯ, ассоциированной с введением гормона роста, сходны и характеризуются церебральной атаксией, слабовыраженной деменцией, лишь в терминальной фазе заболевания выходящей на первый план. Инкубационный, или латентный, период (с момента заражения до первых клинических проявлений) может значительно колебаться — от 2 до более чем 10 лет. Установлена интересная закономерность: деменция развивалась гораздо раньше — уже через 2 года в случае заражения — при пересадке роговицы или твердой мозговой оболочки, нежели при заражении через контаминированный нейрохирургический инструмент [22]. Во всех случаях отмечается массивное отложение амилоида, а также разрастание астроглии во многих областях головного мозга, создающее впечатление «губки». Однако мутации PrP-гена не были установлены абсолютно у всех пациентов, что может быть связано с разнообразием и многовариантностью генотипов PrP-гена и наличием сходных мутаций в нормальной популяции [23].
Новый вариант БКЯ в настоящее время еще называется человеческой формой коровьего губкообразного энцефалита [20]. Он не имеет связи с наследственной формой БКЯ и при нем не выявлены мутации PrP-гена. Это заболевание распространено во всем мире и спорадически встречается на протяжении последнего десятилетия в различных, не связанных между собой регионах, отличается поражением лиц молодого возраста, отсутствием энцефалографических изменений и типичных невропатологических проявлений, характерных для ятрогенной БКЯ [20]. Предвестником данного заболевания являются изменения со стороны психики, такие как девиантное поведение, панические атаки, депрессия. Это может продолжаться от нескольких недель до нескольких месяцев, пока к этим симптомам не присоединятся такие неврологические изменения, как атаксия, а затем и миоклонические судороги. Уже на этом фоне присоединяется прогрессирующая деменция вплоть до развития мутизма. Нейропатологические изменения характеризуются спонгиоформными изменениями в головном мозге — в основном в таламусе и базальных ганглиях: исчезновением нейронов, разрастанием астроцитов. В дополнение к перечисленному в мозговой ткани отмечается отложение амилоидной массы, содержащей PrP-белок. Зачастую молодой возраст этих пациентов в сочетании с характерными клиническими проявлениями, а также связь с пребыванием на территории Великобритании заставляют заподозрить у пациента новый вариант БКЯ.
Спорадическая трансмиссивная губкообразная энцефалопатия
Спорадическая болезнь Крейцфельда — Якобса является наиболее распространенной из всех ТГЭ. В среднем в год регистрируется 1 на 2 × 106 случаев этого заболевания во всем мире [21]. Клинические проявления и патоморфологические изменения при спорадической БКЯ менее вариабельны, нежели в предыдущих двух группах прионных заболеваний [24]. Заболеваемость среди мужчин и женщин приблизительно одинакова. Средний возраст первых клинических проявлений приходится на 50–70 лет, но зарегистрированы единичные случаи от 16–20, максимально — до 80 лет. Основным клиническим симптомом является неуклонно прогрессирующая деменция, характеризующаяся нарастающими когнитивными расстройствами, быстрым снижением памяти и эксцентричным, неадекватным поведением. Постепенно, особенно на поздних стадиях заболевания, присоединяются миоклония, атаксия, пирамидные и экстрапирамидные расстройства, нистагм, диплопия, головная боль, расстройства чувствительности. Энцефалограмма при спорадической БКЯ носит специфический характер. У этих пациентов отмечаются синхронные разряды, состоящие из периодически возникающих трехфазных волн с частотой 1–2 цикла в секунду. Подобные изменения не встречаются в электроэнцефалограмме при семейной и инфекционной БКЯ. Появление подобных изменений при спорадической БКЯ, как правило, является предвестником летального исхода, так как смерть пациентов наступает через 1, максимум 12 месяцев после появления характерных изменений в электроэнцефалограмме. Патоморфологические изменения состоят в разрастании астроглии и спонгиоформных изменениях на фоне исчезновения нейронов, в основном в сером веществе головного мозга. PrP-белок, как правило, обнаруживается в гомогенате, полученном из мозговой ткани. В 5 % случаев был найден PrP-белок в амилоидных отложениях [24].
Семейная трансмиссивная губкообразная энцефалопатия
Семейная ТГЭ (СТГЭ) ассоциируется с присутствием аутосомного доминантного генетического повреждения PrP-гена. В зависимости от характера генетического повреждения клиническая картина заболевания соответствует либо семейной БКЯ, либо синдрому Герстмана — Штраусслера — Шейнкера. Для первого заболевания характерны манифестация и прогрессирование деменции, для второго — церебральная атаксия. Имеющиеся различия в течении и продолжительности заболевания, клинических проявлениях находятся в прямой зависимости от характера мутаций PrP-белка (табл. 2).
/81/81.jpg)
В то же время остается открытым вопрос о возможной роли других дополнительных факторов в манифестации этой группы заболеваний. К таким факторам следует отнести особенности питания, возраст, наличие каких-то экзогенных вирусных агентов и так далее. Вариабельность клинических проявлений различных форм СТГЭ остается загадкой, хотя имеющиеся факты свидетельствуют о наличии корреляции между локализацией отложений PrP в головном мозге и характером клинических проявлений, а также о тяжести течения заболевания. Например, локализация отложения PrP в синапсах в основном наблюдается среди пациентов со спорадической и семейной болезнью Крейцфельда — Якобса с мутацией Lys200, что сопровождается тяжелой деменцией, минимальной атаксией и скоротечностью течения с быстрым наступлением смерти. В противоположность этому у пациентов с куру, ятрогенной ТГЭ вследствие введения фактора роста при гормонотерапии и СТГЭ, ассоциируемой с другими мутациями PrP, заболевание продолжается значительно дольше и сопровождается присоединением выраженной атаксии [25, 26].
Фатальная семейная бессонница
Описана относительно недавно — в 1986 г. Это чрезвычайно редкая прионная болезнь с аутосомно-доминантным типом наследования. На практике такой тип наследования означает, что у носителя этого патологического гена обязательно (рано или поздно) проявится данное заболевание, но степень выраженности болезни может быть различной. Выделяют четыре стадии заболевания.
Первая стадия длится около четырех месяцев. Ее основной признак — прогрессирующая бессонница. На второй стадии, длящейся примерно пять месяцев, присоединяются галлюцинации и тревожно-депрессивный синдром. Третья стадия характеризуется полным отсутствием сна, затем постепенно нарастают признаки деменции (четвертая стадия). Примерно через полгода больные погибают от истощения и/или интеркуррентных воспалительных заболеваний.
Все многочисленные проявления этой болезни обусловлены дистрофическими изменениями таламуса — снижением эффективности проведения импульсов через таламус, нарушением циркадных ритмов, влияющих на кровяное давление, частоту сердечных сокращений, температуру тела и гормональные ритмы, снижением порога болевой чувствительности и рефлекторной активности, развитием слабоумия.
Лабораторная диагностика прионных болезней
1. Для выявления возбудителей прионных болезней в настоящее время разработано несколько методик, в том числе гистологическая и гистохимическая. Материалом для исследования служат гистологические препараты из биоптатов головного мозга, в которых выявляют астроцитоз, губкообразные изменения и отсутствие инфильтративных воспалительных реакций. При гистохимическом исследовании микропрепаратов из биоптата мозга обязательно наличие в них амилоидных отложений [27].
2. Исследование спинномозговой жидкости с целью выявления в ней белковых маркеров возбудителей прионных заболеваний с помощью метода иммуноблоттинга и метода «realtime quaking-induced conversion» ПЦР [27, 28].
3. Генетический анализ прионного гена [29].
Профилактика заражения прионными болезнями
Аминокислотная последовательность нормального клеточного белка PrP у разных видов млекопитающих и человека почти не различается, что указывает на возможность передачи прионных заболеваний от животных к людям, в том числе через зараженное мясо, но вероятность передачи этих болезней человеку крайне низка, так как во многих странах вводятся меры по предупреждению потенциальной опасности заражения через продукты питания. Международный зоосанитарный кодекс запрещает не только импорт костной муки, но и ввоз эмбрионов и спермы племенного скота из стран риска (хотя, по некоторым данным, прионная инфекция не может передаваться через сперму). Во всех странах с риском распространения прионных заболеваний существует запрет на производство костной муки из зараженных животных и использование субпродуктов. В Англии, кроме того, исследуется мозг всего рогатого скота. Молоко считается безопасным, хотя в последнее время стали появляться сообщения, будто употребление молока тоже может иметь печальные последствия.
На сегодняшний день известно несколько путей проникновения патогенного приона в организм человека: интрацеребральный, интравенозный, интраперитонеальный, подкожный, пероральный.
Для обезвреживания инструментов и объектов окружающей среды рекомендуются автоклавирование (134 °С — 18 минут; 121 °С — 1 час), сжигание подозрительного биологического материала, дополнительная обработка отбеливателем и однонормальным раствором NaOH в течение часа. Неспецифическая профилактика включает ограничения на использование лекарственных препаратов животного происхождения без их соответствующей обработки и запрет на применение гормонов гипофиза животного происхождения. Ограничивают также трансплантацию твердой мозговой оболочки и роговицы. При работе с биологическими жидкостями больных используют резиновые перчатки.
Особое место занимают так называемые биопрепараты, которые получают от различных животных. Прежде всего следует обращать внимание на размер входящих в состав биопрепаратов белковых молекул, так как размер прионов колеблется от 33 до 35 кД [4]. Поэтому все препараты, содержащие белковые молекулы меньше этих размеров, не представляют опасности в качестве источника заражения. В частности, такой широко распространенный препарат, как Актовегин, получаемый из крови молодых телят и подвергающийся обработке с получением пептидов с молекулярной массой не более 5 кД, естественно, не может содержать инфекционный прионный агент. Актовегин представляет собой депротеинизированный апирогенный и неиммуногенный гемодиализат телячьей крови. Он готовится посредством нескольких ступеней ультрафильтрации. На первом этапе используется порог в 5 кД, после чего проводятся вакуумная дистилляция, удаление преципитата путем фильтрации через поры диаметром 0,45 мкм и доведение pH до 6,8. После этого полученный продукт фильтруется в стерильных условиях через фильтры с порами 0,2 и 0,45 мкм и выдерживается при t 2–6 °С не менее 14 суток, после чего снова подвергается фильтрации (0,45 мкм) и доведению pH до 6,8. После последующих коррекций pH продукт снова фильтруется (7 и 0,2 мкм) и подвергается еще одной ультрафильтрации с порогом 10 кД и фильтрации в стерильных условиях через фильтры с порами 0,2 и 0,45 мкм. После еще одной выдержки (не менее 56 суток при 2–6 °С) готовый продукт отделяют фильтрацией (0,45 мкм) и разводят до номинальной концентрации 200 мг/мл сухого веса. Депротеинизация завершается фильтрацией через фильтры с порами 0,2 и 0,45 мкм в стерильных условиях.
Анализ готового препарата показывает, что он представляет собой смесь природных веществ, как неорганических — электролитов (хлориды, натрий, калий, кальций, магний, соединения азота), так и органических (глюкоза, ацетат, лактат, аминокислоты, пептиды, нуклеозиды, гликосфинголипиды, другие продукты метаболизма). Поскольку белки удаляются из дефибринированной крови при ультрафильтрации (порог — 5 кД), электрофорез в полиакриламидном геле не обнаруживает в нем следов белка.
Следовательно, при решении вопроса о возможной опасности применения того или иного биопрепарата следует прежде всего обращать внимание на молекулярные характеристики входящих в его состав агентов и методы обработки нативного материала, послужившего основой для его создания.
Впервые опубликовано
в журнале «Фарматека», 2012
ACTO-PUB-092014-067
1. Покровский В.И., Киселев О.И., Черкасский Б.Л. Прионы и прионные болезни. — М., 2004. — 384 с.
2. Gaidusek D.C. Slow-virus infections of the nervous system // N. Engl. J. Med. — 1967. — 287. — 392–400.
3. Prusiner S.B. Novel proteinaceous infectious particles cause scrapie // Science. — 1982. — 216. — 136–44.
4. Liao Y.-C., Lebo R.V., Clawson G.A. et al. Human prion protein: molecular cloning, chromosomal mapping and biological implication // Science. — 1986. — 233. — 364–67.
5. Prusiner S.B. Prions // Proc. Nat. Acad. Sci., USA. — 1998. — 95. — 3363–83.
6. Safar J.G. Molecular pathogenesis of sporadic prion diseases in man // Prion. — 2012. — 2. — 108–15.
7. Prigent S., Rezaer H. PrP assembles // Prion. — 2011. — 5(2). — 68–75.
8. Зуев В.А. Прионы — новый класс возбудителей инфекционных заболеваний // Антибиотики и химиотерапия. — 1999. — № 10. — С. 33–8.
9. Collinge J., Sidle K.C., Meads J. et al. Molecular analysis of prion strain variation and the aethiology of new variant CJD // Nature. — 1996. — 383. — 685–90.
10. Ryon C. Prions and prion diseases: Fundamentals and Mechanistic details // J. Microbiol. Biotechnol. — 2007. — 17. — 1059–70.
11. Pan K.-M., Baldwin M., Nguyen J., Gasset et al. Conversion of alf-helices into beta-sheets features in the formation of the scrapie prion proteins // Proc. Nat. Acad. Sci., USA. — 1993. — 90. — 1062–66.
12. Safar J., Roller P.P., Gajdusek et al. Conformation, transition, dissociation and unfolding of scrapie amyloid (prion protein) // J. Biol. Chem. — 1993. — 268. — 20276–84.
13. Smirnovas W.K. Distinkt structures of scrapie prion protein — seeded versus spontaneous recombinant prion protein fibrils reveald by hydrogen deuterium exchange // J. Biol. Chem. — 2009. — 284. — 2423–41.
14. Griffith J.J. Self replication and scrapie // Nature. — 1967. — 215. — 1043–44.
15. Come J.H., Feaser P.E., Lansburg P.T. A kinetic model for amyloid formation in the prion diseases importance of seeding // Proc. Nat. Acad. Sci., USA. — 1993. — 90. — 5959–63.
16. Makarova N., Baskakov I.V. Genesis of transmissible protein via deformed templating // Prion. — 2012. — 6(3). — 252–55.
17. Gough K.C., Maddison B.C. Prion transmission // Prion. — 2010. — 4(4). — 275–82.
18. Miller M.W., Williams E.S., Hobbs N.T. et al. Enviromental sources of prion transmission in mule deer // Emerg. Infect. Dis. — 2004. — 10. — 1003–6.
19. Georgsson G., Sigurdarson S., Brown P. Infection agent of sheep scrapie may persist in the environment for at least 16 years // J. Gen. Virology. — 2006. — 87. — 3737–40.
20. Will R.G., Zeidler M., Stewart G.E. et al. Diagnosis new variant of Creutzfeldt–Jakob disease // Ann. Neurol. — 2000. — 47. — 575–82.
21. Will R.G. Epidemiology of Creutzfeldt-Jakob disease // Br. Med. Bull. — 1993. — 49. — 960–70.
22. Guiroy D.C., Yanaghara R., Gajdusek D.C. Localization of amy loidogenic proteins and sulfatedglycosaminoglycans in nontransmissible and transmissible cerebral amyloidoses // Acta Neuropathol. — 1991. — 82. — 87–92.
23. Pocchiari M. Prions and related neurological diseases // Mol. Aspects. Med. — 1994. — 15. — 195–291.
24. Broun P., Gibbs C.J., Rodgers-Johnson P. et al. Human spongioform encephalopathy: the National Instittutes of Health series of 300 cases of experimentally transmitted disease // Ann. Neurol. — 1994. — 35. — 513–29.
25. Tateishi J., Kitamoto T. Developments in diagnosis for prion diseases // Br. Med. Bull. — 1993. — 49. — 971–79.
26. Tateishi J., Kitamoto T. Inherited prion diseases and transmission to rodents // Brain Pathol. — 1995. — 5. — 53–9.
27. Ryuichiro Atasari, Kazunori Sano, Katsuya Satoh et al. Real time quaking-induced conversion. A highly sensitive assay for prion detection // Prion. — 2011. — 5(3). — 150–53.
28. Orru Ch.D., Wilham J.M., Hughson A.G. et al. Human variant Creutzfreldt-Jacob disease and sheep screpie PrPres detection using seeded conversion of recombinant prion protein // Prot. Eng. Des. Sel. — 2009. — 22(8). — 515–24.
29. Lukic A., Mead S. Genome wide association studies and prion disease // Prion. — 2011. — 5(3). — 154–60.