
Международный эндокринологический журнал 4 (36) 2011
Вернуться к номеру
Симпозиум «Акромегалия: патогенез, клиника, диагностика, методы лечения»
Авторы: Панькив В.И., Украинский НПЦ эндокринной хирургии, трансплантации эндокринных органов и тканей МЗ Украины
Рубрики: Эндокринология
Версия для печати
Проводит: Донецкий национальный медицинский университет им. М. Горького
Рекомендован: эндокринологам, семейным врачам
Определение
Акромегалия — нейроэндокринное заболевание, вы-званное хронической избыточной секрецией гормона роста (соматотропина, СТГ) у лиц с законченным физиологическим ростом и характеризующееся патологическим диспропорциональным периостальным ростом костей, хрящей, мягких тканей, внутренних органов, а также нарушением функционального состояния сердечно-сосудистой, легочной системы, периферических эндокринных желез, обмена веществ.
Гигантизм — нейроэндокринное заболевание, вы-званное хронической избыточной секрецией СТГ у лиц с незаконченным физиологическим ростом (детей и подростков), характеризующееся пропорциональным ростом костей скелета в длину, приводящее к значительному увеличению линейного роста субъекта. Если такие больные не подвергаются своевременному лечению, то после завершения пубертатного периода у них развиваются все типичные симптомы акромегалии.
Эпидемиология
Чаще всего заболевание акромегалией отмечается между 20 и 40 годами, но наблюдается ее развитие как в возрасте старше 50 лет, так и в возрасте между 10 и 15 годами. На 1 миллион населения приходится от 50 до 70 случаев заболеваний акромегалией. Ежегодно фиксируется 3–4 повторных случая. Однако точную цифру распространенности данного заболевания трудно указать в связи с тем, что время от появления первых признаков акромегалии до установления точного диагноза колеблется от 5 до 15 лет. Не выявлено четкой взаимосвязи частоты возникновения акромегалии с полом, хотя многие авторы указывают на большую предрасположенность женщин к данному заболеванию. При анализе анамнестических данных более половины больных отмечают появление признаков заболевания на фоне полного здоровья, около 18 % больных связывают начало заболевания с предшествующей черепно-мозговой травмой, около 5 % женщин — с повторными абортами и родами, у 20 % больных акромегалией в анамнезе хронические синуиты и отиты с частыми обострениями. Акромегалия характеризуется прогрессирующей инвалидизацией и сокращением продолжительности жизни. Смертность среди больных акромегалией превышает в десять раз таковую в контрольной популяции. Приблизительно 50 % нелеченных больных умирают в возрасте до 50 лет. Основными причинами повышенной смертности и сокращения продолжительности жизни являются осложнения, развивающиеся при данном заболевании: сердечно-сосудистая патология, сахарный диабет и его осложнения, заболевания органов дыхания, злокачественные новообразования желудочно-кишечного тракта и некоторые другие. В свою очередь, своевременная диагностика и адекватное лечение данного заболевания позволяет сократить частоту смертности в 2–5 раз.
Классификация
Наиболее распространена классификация по этиологическому принципу (см. «Этиология и патогенез»). Кроме того, по степени активности выделяют:
— активную стадию (фазу) заболевания;
— стадию (фазу) ремиссии.
По характеру течения выделяют:
— прогрессирующее;
— торпидное.
Этиология и патогенез
В настоящее время предполагается существование следующих основных этиопатогенетических механизмов хронической избыточной продукции гормона роста.
В 95 % случаев это первичная избыточная секреция гормона роста аденомой гипофиза. Гистоморфологический анализ таких аденом показал, что они могут представлять собой плотно- или редкогранулированную соматотропную аденому, смешанную аденому из соматотрофных и лактотрофных клеток, маммосоматотропиному, плюригормональную аденому, а также карциному из соматотрофных клеток.
В настоящее время считается, что развитие аденом гипофиза является многоступенчатым процессом, в котором первичные генетические изменения вызывают нерегулируемую моноклональную экспансию мутированных клеток. Гипоталамические гормоны и локальные ростовые факторы играют роль промоторов дальнейшего роста. Конечный фенотип опухоли гипофиза является результатом взаимодействия активизирующих и тормозящих генетических мутаций с локальными гипоталамическими эндокринными и паракринными ростовыми факторами.
К активизирующим клеточным мутациям относится активация клеточных протоонкогенов. Протоонкоген — нормальный клеточный ген, вовлеченный в контроль клеточной пролиферации и дифференцировки, который в случае мутации активизируется, что может приводить к опухолевой трансформации. Мутант-ные аллели протоонкогенов являются доминантными и способны трансформировать клетки, несмотря на одновременную экспрессию нормальной аллели. Так, в 40 % СТГ-продуцирующих аденом гипофиза была обнаружена мутация альфа-субъединицы G-белка — самая частая мутация при данном типе аденом. При этом данная мутация очень редко встречается при других типах опухолей гипофиза: НАГ — < 10 %, кортикотропиномы — < 6 %. Частота этой мутации имеет также этническую зависимость: наиболее редко она встречается в японской популяции (менее 10 %). Образованный в результате мутации gsp-онкоген вызывает подавление активности гуанозинтрифосфата на альфа-субъединице аденилатциклаззависимого стимулирующего G-белка. Это приводит к персистирующей Gs-активации аденилатциклазы, которая имитирует постоянную активацию рецепторов соматолиберина и приводит к автономной секреции СТГ и гиперплазии соматотрофов (рис. 1).
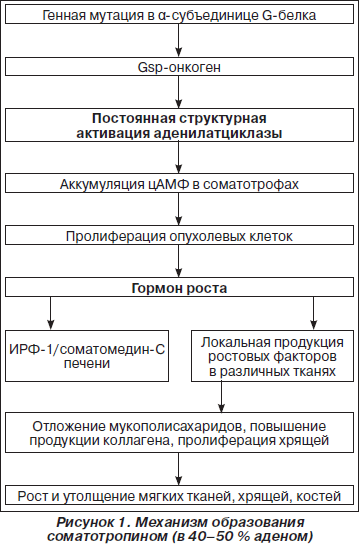
Считается, что постоянной активации аденилатциклазы способствует возникающий в рецепторах, экспрессируемых в соматотрофах, трансдукционный дефект, приводящий к утере ингибирования аденилатциклазы.
Исследования не выявили корреляции между присутствием gsp-мутации и возрастом, полом, размером опухоли или циркулирующим уровнем СТГ и ИРФ-1. Gsp-экспрессирующие опухоли по данным морфологического анализа являются плотногранулированными, слабоинвазивными, наименее агрессивными СТГ-секретирующими опухолями, чрезвычайно чувствительными к подавляющему эффекту соматостатина.
В СТГ-продуцирующих аденомах гипофиза с меньшей частотой обнаружена повышенная активность и других клеточных протоонкогенов, в частности, протеинкиназы С (РКС) — фермента семейства кальций- и фосфолипидзависимых белковых киназ. РКС, являясь важным энзимом в передаче клеточных сигналов в гипофизе, стимулирует форболовые эстеры, способствующие опухолевому развитию. Более того, инвазивный рост опухоли сочетается с обнаружением мутации V3 области альфа-изоформы РКС.
Не исключена также роль в гипофизарном туморогенезе мутаций мощного онкогена — гена, трансформирующего опухоль гипофиза (PTTG). Повышение экспрессии данного онкогена более чем на 50 % отмечено в большинстве СТГ-секретирующих аденом гипофиза, а самая высокая экспрессия — в активных опухолях с инвазией в клиновидную кость. Повышение экспрессии PTTG приводит к нарушению разделения хромосом, ведущему к потере или появлению избыточной хромосомы. Последующая хромосомная анеуплоидия может приводить к активации протоонкогенов или потере гетерозиготности опухоль-супрессорных генов.
Опухоль-супрессорные гены в норме ограничивают клеточную пролиферацию, регулируя клеточный цикл и поддерживая стабильность генома, таким образом пред-отвращая манифестацию опухолевого роста. Инициация роста опухолевой клетки начинается после того, как обе рецессивные аллели опухоль-супрессорного гена утеряны или изменены. Среди опухоль-супрессорных генов, с большой вероятностью играющих роль в генезе гипофизарных опухолей, в частности, соматотропином, выделяют ген множественной эндокринной неоплазии 1, ген ретинобластомы и некоторые из генов CDK1.
Очень редко (< 2 %) гиперфункция гормона роста может быть вызвана СТГ-секретирующей опухолью внегипофизарной локализации. Последняя может быть обнаружена в глотке, поджелудочной железе, легких, яичниках, средостении. Механизм ее возникновения предположительно аналогичен СТГ-продуцирующей аденоме гипофиза.
Менее чем в 3 % всех случаев акромегалия возникает вследствие повышенной секреции соматолиберина, приводящей к гиперплазии соматотрофов с последующим формированием поликлональной аденомы гипофиза. Такая патологическая избыточная секреция соматолиберина может возникнуть в случае развития как опухолей гипоталамуса (гамартома, ганглионеврома), так и эктопированных соматолиберин-продуцирующих опухолей. Последние могут быть представлены карциноидом, расположенным в бронхах, желудочно-кишечном тракте, поджелудочной железе; опухолью островков Лангерганса, а также мелкоклеточным раком легкого.
Наконец, предполагается существование безопухолевой гиперсекреции гипоталамусом соматолиберина вследствие воспалительных процессов в ЦНС (арахноидит и др.) как возможной причины развития акромегалии.
Продуцируемый в избытке гормон роста индуцирует повышенную секрецию ростовых факторов (соматомединов), в основном ИРФ-1 (соматомедина С), который вырабатывается в печени, а также локальную продукцию ростовых факторов в различных тканях, включая кости и хрящи. Под воздействием ростовых факторов в мягких тканях происходит отложение мукополисахаридов, включая глюкозаминогликаны, гиалуроновую кислоту и хондроитин-сульфат; возникает повышение продукции коллагена, пролиферация хрящей, что в конечном итоге приводит к росту и утолщению мягких тканей, хрящей и костей и объясняет появление главных клинических симптомов заболевания. Последние исследования указывают на весьма вероятную роль местной продукции ИРФ-связывающих белков, в частности белка-3, в стимуляции остеобластов и хондроцитов (рис. 1).
Акромегалия и гигантизм могут быть вызваны:
— опухолью гипофиза (95 %): аденомой гипофиза (соматотрофной, маммосоматотропиномой, ацидофильной стебельно-клеточной, плюригормональной); аденокарциномой;
— эктопической секрецией гормона роста (менее 2 %): эндокраниальной (опухоль глоточного и сфеноидального синуса), экстракраниальной (опухоль поджелудочной железы, легких, средостения);
— эктопической секрецией соматолиберина (менее 3 %): эндокраниальной (гамартома, ганглиоцитома), экстракраниальной (карциноид поджелудочной железы, бронхов, ЖКТ);
— в рамках наследственных синдромов: синдром Мак-Кьюна — Олбрайта (McCune — Albright), синдром МЭН-1, семейная акромегалия, синдром Карни (Саrny).
По размеру выявляемой аденомы гипофиза выделяют:
1) микроаденому (менее 10 мм);
2) макроаденому (более 10 мм):
— эндоселлярная (мезоаденома);
— экстраселлярная: параселлярная (латероселлярная), супраселлярная (со зрительными нарушениями и без них), инфраселлярная, гигантская.
Клиника
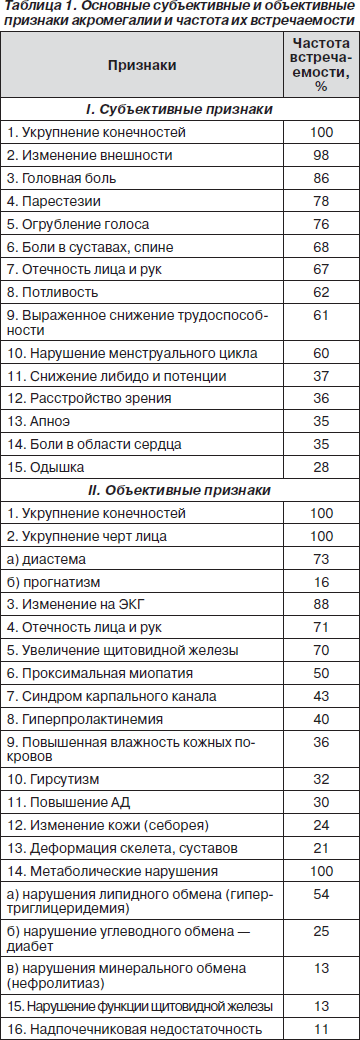
Клиническая картина акромегалии весьма много- образна, что обусловлено вовлечением в патологический процесс многих органов и систем при данном заболевании (табл. 1). Наиболее ярким клиническим проявлением акромегалии является изменение внешности. Так, больные отмечают укрупнение носа, губ, языка, утолщение кожи, надбровных дуг. Возникает диастема (расширение межзубных промежутков), прогнатизм (выстояние нижней части лица), особенно нижней челюсти, что приводит к нарушению прикуса. Увеличиваются в размерах конечности: кисти, пальцы, стопы, причем в основном в ширину, что вынуждает больных менять перчатки, кольца, обувь. В укрупненных конечностях часты парестезии, онемение пальцев. У 35–43 % больных развивается синдром карпального канала вследствие сдавления срединного нерва в области запястья измененными мягкими тканями.
Характерным симптомом является головная боль, нередко постоянная, изнуряющая, частота которой составляет 60–86 %. Оказываемое опухолью давление на окружающие ткани не является единственной причиной головных болей. Генез последних до сих пор полностью не изучен. Повышенная активность потовых и сальных желез приводит к повышенной потливости, появлению жирной себореи, acne vulgaris, что обусловливает специфический неприятный запах тела. Возникают изменения в периферических нервах, в частности сегментарная демиелинизация нервных волокон малого диаметра, что подтверждается результатами биопсии. Этот процесс, скорее всего, обусловливает появление таких клинических симптомов, как парестезии, а также снижение периферических рефлексов, поверхностных тактильных и болевых ощущений.
Несмотря на гиперфункцию мышечных волокон, приводящую к увеличению мышечной массы, многих больных беспокоит быстрая утомляемость, слабость, резко снижается трудоспособность, толерантность к физическим нагрузкам. Последняя связана с развивающейся почти у 50 % больных проксимальной миопатией, подтверждаемой результатами электромиографии.
Основные клинические симптомы и осложнения акромегалии:
I. Костная система
1. Диастема
2. Прогнатизм
3. Фронтальный гиперостоз
4. Заболевания височно-нижнечелюстного сустава
5. Остеоартриты
6. Дорзальный кифоз
II. Кожа
1. Akantosis nigricans
2. Грубые кожные складки
3. Аспе
4. Гирсутизм
5. Профузная потливость
6. Себорея
7. Бородавки
8. Гидраденит
III. Эндокринная система и метаболические нарушения
1. Нарушения менструального цикла
2. Снижение либидо и потенции
3. Лакторея с гиперпролактинемией и без таковой
4. Узлы щитовидной железы с нарушением функции и без таковой
5. Гипертриглицеридемия
6. Нарушение толерантности к глюкозе и диабет
7. Гиперкальциурия с уролитиазом
8. Холелитиаз
IV. Центральная и периферическая нервная система
1. Сужение полей зрения
2. Парез черепно-мозговых нервов
3. Синдром карпального канала
4. Проксимальная миопатия
5. Радикулопатия
V. Сердечно-сосудистая система
1. Артериальная гипертензия
2. Кардиомиопатия (гипертрофия левого желудочка, нарушения сердечного ритма)
3. ИБС
4. Нарушения мозгового кровообращения
VI. Система органов дыхания
1. Ночные апноэ (обструктивные и центральные)
2. Рестриктивные заболевания
VII. Онкологические заболевания
1. Аденоматозные полипы
2. Рак толстой кишки
Приблизительно 62–75 % больных беспокоят боли в суставах, позвоночнике по типу радикулярных, что связано с развитием артропатии, вплоть до остеоартритов, а также деформации скелета по типу патологического кифоза. Избыточная продукция коллагена внутри сухожилий вызывает дополнительную дестабилизацию суставов. Постоянная стимуляция остеобластов ведет к дегенеративным изменениям в околосуставных областях. Рентгенографически при этом выявляются увеличение размера суставного пространства (в поздних стадиях — уменьшение), угловые деформации суставов, остеофиты, обызвествление суставных поверхностей, остеосклероз, субхондральные кисты, увеличение и кальцификация реберно-хрящевых сочленений. Утолщение голосовых связок и расширение синусов приводит к появлению низкого грубого голоса. У женщин нередко появляется гирсутизм, лакторея; у 32–87 % из них возникают нарушения менструального цикла, чаще по типу олиго- и аменореи; 27–46 % мужчин отмечают снижение либидо и потенции. У 4,0–5,2 % больных отмечается нарушение зрения, проявляющееся как снижением остроты, так и ограничением полей, вплоть до полной слепоты. Появление этих симптомов характерно для больших аденом гипофиза и обусловлено супраселлярным ростом опухоли, которая сдавливает перекрест зрительных нервов, расположенных на передней поверх-ности гипофиза. В связи с вышеизложенным больным показано офтальмологическое обследование, включающее периметрию, что позволяет выявлять моно- или битемпоральную гемианопсию с центральной скотомой или без таковой, атрофию зрительных нервов, зрачковые дефекты.
Значительное распространение опухоли гипофиза латерально в кавернозные синусы может привести к нарушению функции (парезу) III, IV, V и VI пар черепно-мозговых нервов, части которых проходят в кавернозных синусах. Клинически это может проявиться офтальмоплегией, птозом, дисфункцией зрачков, болями по ходу тройничного нерва, снижением рефлексов. При значительном супраселлярном распространении опухоли возможна обструкция III желудочка, приводящая к повышению внутричерепного давления и появлению отека соска зрительного нерва.
Одним из серьезных осложнений при акромегалии являются изменения со стороны сердечно-сосудистой системы. Так, у 25–50 % больных фиксируется артериальная гипертензия, что в 3–4 раза чаще, чем в популяции. Генез развития артериальной гипертензии при акромегалии изучен недостаточно, однако считается, что основным предрасполагающим фактором является задержка натрия и воды в организме. Характерным является увеличение сердца и развитие сердечной недостаточности. Кардиомегалия является одним из проявлений спланхномегалии, характерной для данного заболевания. Масса миокарда прямо пропорциональна длительности заболевания и может достигать 1300 г. Причиной развития кардиомегалии являются концентрическая гипертрофия сердечной мышцы и повышенный синтез соединительной ткани.
Развитию гипертрофии миокарда, особенно левого желудочка способствует также артериальная гипертензия.
Со временем в гипертрофированном миокарде развиваются дистрофические изменения, приводящие к развитию так называемой акромегалической кардиомиопатии, что подтверждается появлением изменений на электрокардиограммах, а также результатами ауто-псии. В частности, у более 88 % больных независимо от возраста выявляются диффузные изменения миокарда, нарушения кровоснабжения, нарушения сердечного ритма вплоть до полного A-V-блока. Гистологическое исследование миокарда умерших больных в 93 % случаев выявляет гипертрофию миокарда, в 85 % — интерстициальный фиброз и в 59 % случаев — лимфомононуклеарный миокардит.
Развитию дистрофии миокарда способствуют развивающиеся при акромегалии метаболические нарушения, микроангиопатия, гипоксия и т.д. Поражение сердца часто усугубляется присоединением атеросклероза коронарных артерий, что встречается до 20 %. Постепенно прогрессируя, миокардиодистрофия приводит к развитию сердечной недостаточности и нередко является причиной летального исхода.
Поражение легочной системы также является серьезной проблемой больных акромегалией. Вследствие разрастания челюстей и мягких тканей языка и надгортанника у 60 % больных, в основном мужчин, развиваются обструктивные ночные апноэ. Прогрессирующий кифосколиоз приводит к развитию рестриктивных легочных заболеваний. В итоге смертность от нарушения дыхания у больных с акромегалией превышает таковую в 3 раза по сравнению с контрольной популяцией.
И наконец, частота новообразований у больных акромегалией превышает таковую в общей популяции более чем в 2 раза. Результаты биопсии свидетельствуют о преобладании кишечных аденоматозных полипов, а также рака толстой кишки.
Хроническая избыточная секреция гормона роста приводит также к выраженным метаболическим сдвигам. К ним относятся развитие нарушений углеводного, липидного и минерального обменов.
Частота нарушений углеводного обмена достигает 54 %, на долю диабета при этом приходится до 25 %. Главной отличительной особенностью диабета при акромегалии является резистентность к традиционным методам лечения и, в частности, к инсулинотерапии.
Исследования показали, что степень повышения гормона роста, длительность заболевания, возраст больных, наследственная предрасположенность, особенности HLA-фенотипа не влияют на частоту и степень нарушений углеводного обмена. Установлено также, что последние не связаны с истощением секреторной способности b-клеток, гиперпродукцией глюкагона, а также нарушением функционирования инсулин-специфических рецепторов. Предполагается, что в основе нарушений углеводного обмена при акромегалии лежат изменения в центральной регуляции секреции инсулина и глюкагона в сочетании с нарушением белкового транспорта глюкозы на пострецепторном уровне, а также с нарушением образования метаболически активных форм инсулина.
У всех больных акромегалией обнаруживаются сдвиги в липидном обмене, положительно коррелирующие со степенью нарушения углеводного обмена. В частности, отмечается сдвиг липопротеидного спектра плазмы в сторону a-липопротеидов, более высокое содержание в крови холестерина, НЭЖК, кетоновых тел и особенно триглицеридов. Уровень последних статистически достоверно отличается от нормы. Появление гипертриглицеридемии объясняется снижением активности печеночной триглицерид- и липопротеинлипазы.
Изменения в минеральном обмене характеризуются нарушением фосфорно-кальциевого обмена. В частности, возникает повышенная реабсорбция фосфора в канальцах почечных нефронов, что приводит к гиперфосфатемии приблизительно у 48 % больных. Повышение активности 1-a-гидроксилазы в почках вызывает повышение в крови уровня 1,25 гидроксивитамина D и кальция, что, в свою очередь, способствует камнеобразованию в почках у 6,0–12,5 % больных.
Повышение секреции СТГ аденомой гипофиза может вызвать нарушение функций других отделов аденогипофиза, в частности гонадотрофов, кортикотрофов и тиреотрофов, что, в свою очередь, приводит к нарушению функционального состояния периферических эндокринных желез: гонад, щитовидной железы и надпочечников.
Как отмечено выше, у женщин, страдающих акромегалией, часто возникают нарушения менструального цикла, бесплодие. При этом у 2/3 больных выявляется лакторея. Последняя в значительном проценте случаев встречается и у больных акромегалией мужчин.
Основной причиной вышеописанных симптомов является гиперпролактинемия, выявляемая в 40–84 % случаев. Последняя является также основной причиной снижения либидо и потенции у мужчин, больных акромегалией. При этом у части женщин с гиперпролактинемией возможно отсутствие лактореи, что объясняется низкими уровнями эстрогенов, необходимых для начала процесса лактации. Наличие же лактореи у женщин с нормальным уровнем пролактина, вероятно, обусловлено пролактиноподобным действием гормона роста.
Появление гиперпролактинемии может быть обусловлено различными причинами, основной из которых является наличие смешанной аденомы гипофиза, состоящей из соматотрофных и лактотрофных клеток, секретирующих СТГ и пролактин (ПРЛ), либо маммосоматотропиномы. Как следствие гиперпролактинемии отмечается достоверное снижение уровня гонадотропинов и тестостерона. При отсутствии своевременного лечения постепенно развивается клиническая картина вторичного гипогонадизма.
В 30–97 % случаев обнаруживается увеличение щитовидной железы с узлообразованием и без такового, что объясняется прямым стимулирующим действием ИРФ-1 на ткань железы. Гистологический анализ указывает на преобладание (69 %) коллоидного пролиферирующего зоба. При этом имеют место также аутоиммунный тиреоидит, папиллярная цистаденома, микрофолликулярная аденома.
В большинстве случаев структурные изменения в железе сопровождаются эутиреоидным состоянием. Клиника гипотиреоза, подтвержденная лабораторными данными, развивается либо в случае массивного узлообразования, особенно на фоне аутоиммунного тиреоидита (первичный гипотиреоз), либо при сдавлении опухолью гипофиза тиреотрофов, приводящем к снижению секреции тиреотропного гормона (ТТГ) (вторичный гипотиреоз). Возможно и сочетание этих процессов.
Ряд ученых объясняют снижение секреции ТТГ повышением секреции соматостатина гипоталамусом, индуцируемого высоким уровнем СТГ.
Тиреотоксикоз при акромегалии наблюдается редко. Так, частота первичного тиреотоксикоза, по данным различных авторов, колеблется от 2 до 10 %. Описаны единичные больные с вторичным тиреотоксикозом, являющимся следствием гиперпродукции ТТГ смешанной аденомой гипофиза.
Функция надпочечников у больных акромегалией, как правило, не нарушается. Однако, по литературным данным, до 20 % больных могут иметь вторичную надпочечниковую недостаточность вследствие сдавления опухолевой массой кортикотрофов и снижения секреции АКТГ при больших, чаще всего экстраселлярных соматотропиномах. При этом в первые годы заболевания возможно развитие симптомов гиперфункции с последующим истощением коры надпочечников и развитием явлений гипокортицизма.
При морфологическом исследовании в надпочечниках отмечается гиперплазия клеток коркового вещества, в некоторых случаях — аденомы и кистозные перерождения. Крайне редко возможно сочетание акромегалии и болезни Иценко — Кушинга.
Диагноз и дифференциальный диагноз
Как следует из самого названия заболевания, наиболее ярким клиническим проявлением акромегалии является изменение внешности, и при наличии у больного характерных жалоб диагноз часто не вызывает сомнений. Однако ограничиваться результатами клинического обследования абсолютно недостаточно. Только полное лабораторное и инструментальное обследование позволяет правильно поставить диагноз, определить степень активности заболевания и адекватную лечебную тактику.
В диагностический алгоритм акромегалии (рис. 2) включено проведение рентгенографического обследования (рентгенография черепа в боковой проекции, позвоночника, кистей, стоп). Последнее позволяет выявить не только явные или косвенные признаки аденомы гипофиза, но и другие рентгенографические признаки, характерные для данного заболевания.
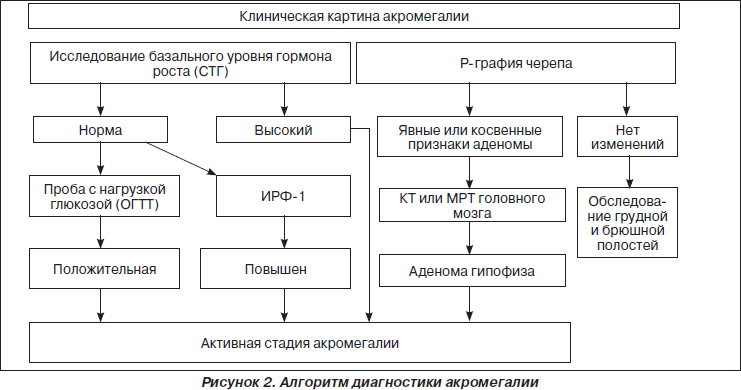
Рентгенографические признаки акромегалии:
I. Аденома гипофиза
1. Явные признаки:
а) увеличение размеров турецкого седла;
б) двухконтурность турецкого седла.
2. Косвенные признаки:
а) локальный или тотальный остеопороз спинки или стенок седла;
б) локальное истончение стенки седла;
в) истечение передних и задних клиновидных отростков;
г) неровность участка внутреннего контура костной стенки седла.
II. Утолщение костей черепа
III. Эндокраниоз
IV. Выраженная пневматизация костей лицевого черепа и пирамид височных костей
V. Гипертрофический остеопороз
VI. ТМТС > 22 мм
Значительные изменения при акромегалии претерпевает структура мягких тканей стопы, что проявляется утолщением соединительно-тканных перегородок и расширением жировых прослоек. В связи с этим важным дополнительным диагностическим рентгенографическим показателем является величина толщины мягких тканей стопы (ТМТС), высоко коррелирующая с уровнем гормона роста. Методика определения данного показателя следующая. Производится боковой снимок стопы. Пучок рентгеновских лучей центрируется на среднюю точку линии, соединяющей медиальную лодыжку с нижней поверхностью стопы. Далее к линии, соединяющей передний и задний отростки пяточной кости, проводится параллельная ей линия касательно к нижнему краю пяточной кости. От точки пересечения этой линии с нижним краем пяточной кости проводится перпендикулярная линия до подошвенной поверхности стопы. Эта линия и является размером ТМТС. Норма у мужчин до 21 мм, у женщин — до 20 мм. В отличие от здоровых лиц данный показатель при акромегалии не зависит от массы тела и возраста. Отсутствует также зависимость и от длительности заболевания акромегалией.
Однако рентгенографическое исследование не позволяет определить размер, характер распространения аденомы гипофиза, степень вовлечения в патологический процесс латеро- и супраселлярных структур, что необходимо знать при определении последующей лечебной тактики. В связи с этим всем больным необходимо проведение компьютерной томографии (КТ) либо магнитно-резонансной томографии (МРТ) головного мозга и области турецкого седла. Как показывает опыт, КТ должна проводиться с обязательным контрастированием. При этом оптимальным методом визуализации аденомы гипофиза, особенно в сочетании с кистозным компонентом, «пустым» турецким седлом, а также в случае микроаденомы, является, безусловно, МРТ. Возможность проведения исследования в трех взаимно перпендикулярных проекциях (сагиттальной, аксиальной и фронтальной) позволяет получить дополнительную информацию об анатомо-топографических особенностях и изменениях селлярной области, более точно определить степень вовлечения в процесс латероселлярных структур. Отсутствие лучевой нагрузки и возможность применения данного метода многократно особенно ценно для динамического наблюдения за опухолями гипофиза на фоне проводимого лечения. Дополнительное использование количественных МРТ-параметров позволяет проводить четкую дифференциацию между пролиферативными процессами в гипофизе и кистозными изменениями, надежно доказать «пустое» турецкое седло.
В настоящее время в мировой практике нашли применение различные диагностические маркеры акромегалии.
Диагностические маркеры акромегалии:
СТГ:
— базальный или случайный;
— каждые 10 мин в течение 24 ч.
Тест с тиролиберином и соматолиберином
ИРФ-1-связывающий белок-3
Соматолиберин в крови
СТГ в крови из нижних кавернозных синусов
СТГ в суточной моче
СТГ в процессе проведения ОГТТ
ИРФ-1 (соматомедин-С)
Основным в лабораторной диагностике является изучение секреции гормона роста (соматотропного гормона, СТГ). Традиционно диагностика акромегалии основывается на выявлении хронически повышенных уровней данного гормона. У большинства больных акромегалией базальные уровни гормона роста колеблются от 5 до 40 нг/мл, в очень небольшом проценте случаев достигая 100–300 нг/мл. При этом от 30 до 53 % больных имеют лишь умеренное или незначительное повышение уровня СТГ. Более того, почти у 17 % больных базальные уровни гормона роста находятся в пределах нормальных значений. Изучение характера секреции СТГ при акромегалии показало, что она отличается выраженной пульсацией: у одного и того же больного можно зафиксировать быстрый подъем в сыворотке крови уровня гормона роста от 5–10 до 50–80 нг/мл в течение 20–40 минут. С другой стороны, многократные измерения показали, что у здоровых лиц также отмечаются подобные пульсации, в частности базальные уровни СТГ колеблются от 0,2 до 20–40 нг/мл (5–10 импульсов в день). Более того, нередко можно видеть пациентов с клиническими признаками активной акромегалии, у которых в случайно взятой пробе крови уровень СТГ будет меньше 1,5 нг/мл. Существует ряд состояний и заболеваний, при которых может быть ложное повышение уровня СТГ натощак. К ним относятся: боль, стресс, инсулинозависимый сахарный диабет, хронические заболевания почек, недоедание, длительное голодание и др.
Причины повышения СТГ, не обусловленные акромегалией:
— боль;
— беременность;
— пубертатный период;
— стресс;
— хронические заболевания почек;
— заболевания сердца;
— сахарный диабет;
— плохое питание, недоедание;
— длительное голодание.
С учетом вышеизложенного однократно полученное значение СТГ не является информативным из-за частого совпадения между здоровыми лицами и больными акромегалией. Иначе говоря, однократное определение базального уровня гормона роста не может как подтвердить, так и исключить диагноз акромегалии, особенно в случаях так называемой «мягкой» акромегалии, когда имеются минимальные клинические проявления заболевания.
Оптимальным является частое (каждые 10–20 минут в течение 24 часов) определение уровня СТГ в крови, что позволяет четко дифференцировать здоровых лиц от больных акромегалией. В норме в 75 % проб содержание гормона роста определяется на уровне нижней границы чувствительности метода, в 25 % проб допускаются высокие значения уровня СТГ (секреторные пики в полночь, ранние утренние часы). В активной стадии акромегалии уровень СТГ в сыворотке крови постоянно повышен в течение 24 часов. Интегрированные суточные уровни гормона роста у каждого больного превышают нормальные значения в 2–100 раз, иногда и более. К сожалению, проведение данного исследования практически очень трудно выполнимо.
В повседневной практике максимальное распространение получило применение орального глюкозо-толерантного теста (ОГТТ), так как прием глюкозы (75 г) вызывает снижение уровня СТГ вплоть до минимально определяемых у 94 % здоровых лиц, но не у больных акромегалией. При проведении теста забор крови проводится натощак, а также каждые 30 минут в течение 2–3 часов после приема глюкозы. При активной стадии акромегалии тест считается положительным, если отсутствует снижение концентрации гормона роста меньше 1 нг/мл. Такая реакция отмечается у большинства больных. Более того, до 30 % лиц с данным заболеванием имеют парадоксальный подъем уровня СТГ в ответ на гипергликемию. Проблема возникает, когда тест проводится у больных с минимально выраженными клиническими признаками акромегалии, базальным уровнем СТГ от 1 до 3 нг/мл. Благодаря техническому прогрессу созданы новые «суперчувствительные» методики, в частности иммунолюминометрический и флюорометрический методы с чувствительностью до 0,005 нг/мл, реально определяющие базальный уровень гормона роста у нормальных лиц. Предварительные результаты у здоровых лиц показали, что у молодых женщин в течение ОГТТ уровень СТГ составляет ниже 0,2 нг/мл, у молодых мужчин — ниже 0,1 нг/мл. Дальнейшее расширение исследований с включением больных с акромегалией в конечном результате позволит использовать ОГТТ для диагностики «мягкой» акромегалии.
В настоящее время показано, что применение ранее широко используемого теста с тиреолиберином (в/в 500 мкг) не несет дополнительной информации в диагностике акромегалии. Более того, тиреолиберин может стимулировать выброс СТГ у многих лиц, не страдающих акромегалией: при декомпенсированном сахарном диабете, голодании, заболеваниях почек и печени, депрессии, психозах, у многих здоровых молодых женщин. Тест рекомендовано применять в послеоперационном периоде при наличии сомнений в радикальности хирургического вмешательства. Получение результатов теста, сходных с дооперационными, может указывать на наличие остаточной аденоматозной ткани.
Тест с соматолиберином (в/в 100 мкг) также не является ценным как в диагностике СТГ-продуцирующих аденом, так и в диагностике эктопической продукции соматолиберина.
Относительно недавно было предложено использовать в качестве нового «интегратора» секреции СТГ значение содержания в крови ИРФ-1-связывающего белка-3, выработка которого индуцируется одновременно гормоном роста и ИРФ-1. Однако в ряде исследований указывается на частичное совпадение результатов между впервые выявленными больными акромегалией и здоровыми лицами. Считается, что обнаружение повышенных уровней ИРФ-1-связывающего белка-3 подтверждает наличие у пациента гиперпродукции СТГ, однако получение нормальных уровней данного белка не исключает диагноза акромегалии. Определение уровня данного белка рекомендуется проводить в пограничных случаях, когда имеется сочетание положительного ОГТТ в отношении подавления СТГ с минимально повышенным уровнем ИРФ-1.
Единственным показанием к определению в сыворотке крови уровня соматолиберина является подозрение на его эктопическую продукцию, которое основывается на отсутствии МРТ-признаков аденомы гипофиза и выявлении объемного образования в грудной либо брюшной полости у пациента с клинической картиной акромегалии. При данном заболевании уровни соматолиберина превышают 300 пг/мл, тогда как во всех остальных случаях, включая и гипоталамическую гиперпродукцию соматолиберина, его уровень составляет менее 50 пг/мл. В литературе представлено 50 случаев акромегалии, обусловленной эктопической продукцией соматолиберина.
Предлагается также использование определения содержания гормона роста в ночной либо суточной моче. Проведенные исследования указывают на хорошую корреляцию между данным показателем и содержанием СТГ и ИРФ-1 в крови. Требуется дополнительная информация в отношении надежности данного маркера. Скорее всего, он станет полезным в качестве скринингового теста либо мониторирования активности заболевания.
Определение содержания СТГ в крови, оттекающей из нижних кавернозных синусов, показано в случаях, когда необходимо уточнение локализации микроаденомы ввиду отсутствия ее четких МРТ-признаков, либо в случае подозрения на гиперплазию гипофиза. Как правило, сочетают с внутривенным введением соматолиберина.
По мнению многих авторов, наилучшим диагностическим маркером, подтверждающим хроническую гиперпродукцию СТГ, является уровень ИРФ-1 в плазме крови. Такое утверждение основывается на ряде факторов.
Преимущества ИРФ-1 как диагностического маркера в сравнении с СТГ:
— ИРФ-1 в конечном итоге ответствен за большинство клинических проявлений акромегалии.
— Уровень ИРФ-1 отражает средний уровень СТГ за предшествующий день ИРФ-1, в отличие от СТГ не подвержен колебаниям в течение короткого периода времени благодаря длительному периоду полужизни.
— Даже незначительно повышенный уровень СТГ сопровождается высоким уровнем ИРФ-1.
Таким образом, в отличие от уровня СТГ однократное определение уровня ИРФ-1 может реально быть использовано для дифференциальной диагностики лиц с нормальной и патологической секрецией СТГ. Для правильной интерпретации получаемых результатов уровня ИРФ-1 необходимо помнить ряд положений:
— связь между гормоном роста и ИРФ-1 носит взаимно логарифмический характер. В частности, при уровне СТГ более 20 нг/мл кривая содержания ИРФ-1 представлена в виде «плато». Возможно, этот уровень СТГ максимально эффективен в отношении способности печени синтезировать ИРФ-1. В определенной мере это позволяет объяснить сходную активность заболевания со значительно различающимися уровнями СТГ;
— уровень ИРФ-1 зависит от пола и возраста, а также от характера питания. Ложное снижение уровня ИРФ-1 (вплоть до нормального значения, соответствующего данному полу и возрасту) возможно при недостаточности питания любого происхождения, голодании, тяжелых заболеваниях печени.
В настоящее время международным консенсусом разработаны четкие критерии исключения акромегалии.
Критерии исключения акромегалии:
— Случайный уровень СТГ < 0,4 нг/мл.
— Нормальный уровень ИРФ-1.
— Минимальный уровень СТГ на фоне ОГТТ < 1 нг/мл (2,7 МЕд/л).
— Средний интегрированный уровень СТГ за сутки < 2,5 нг/мл.
В диагностический алгоритм с целью определения дальней лечебной тактики также входит определение уровня пролактина для решения вопроса о возможности терапии агонистами дофамина; определение функции периферических эндокринных желез для решения вопроса о необходимости проведения заместительной гормональной терапии, а также оценка состояния зрения, как остроты, так и полей.
Дифференциальный диагноз акромегалии должен проводиться с такими заболеваниями, как синдром множественной эндокринной неоплазии (МЭН-1-синдром), гипотиреоз, пахидермопериостоз, болезнь Педжета, синдром McCune-Albright, а также акромегалоидизм.
Клиническая картина акромегалии с развитием аденомы может быть проявлением МЭН-1-синдрома, для которого обязательным является также наличие гормонально-активных опухолей паращитовидных желез, островков поджелудочной железы, а иногда и опухоли легких. Выявление семейных случаев акромегалии также указывает на присутствие МЭН-синдрома.
При гипотиреозе возможны сходные с акромегалией изменения черт лица, кожи, голоса. Диагноз исключается при выявлении снижения функции щитовидной железы в сочетании с нормальными уровнями СТГ.
Редкое семейное заболевание, известное как пахидермопериостоз, может быть ошибочно принято за акромегалию, так как характеризуется грубыми чертами лица, утолщением кожи и гипертрофической остеоартропатией. Выявление нормальных уровней СТГ, как базальных, так и при проведении функциональных тестов, отсутствие признаков аденомы гипофиза отвергает диагноз акромегалии.
При болезни Педжета происходит утолщение проксимальных отделов длинных трубчатых костей, их дугообразное искривление, увеличение черепа за счет утолщения костей свода и основания. Однако отсутствуют характерные для акромегалии изменения мягких тканей, области турецкого седла, определяется нормальное содержание СТГ в крови. Встречается незначительное число лиц с внешними сходными признаками акромегалии в сочетании с нормальным уровнем СТГ. Это так называемый акромегалоидизм.
Для редкого синдрома McCune — Albright, помимо клинической картины акромегалии, характерна также специфическая триада: полиостотическая фиброзная дисплазия, преждевременное половое созревание, специфические пигментные пятна бледно-кофейного цвета.
Рекомендуемые клинические исследования
У преобладающего числа больных причиной заболевания является аденома гипофиза, поэтому обязательными инструментальными методами исследования считаются:
— рентгенография черепа в боковой проекции, позволяющая выявить увеличение размеров турецкого седла (в норме саггитальный размер составляет 12–15 мм, вертикальный — 8–9 мм), его двухконтурность, а также ряд косвенных рентгенографических признаков опухоли гипофиза;
— определение толщины мягких тканей стопы, величина которой выраженно коррелирует с уровнем СТГ (норма у мужчин — до 21 мм, у женщин — до 20 мм);
— КТ головного мозга и области гипофиза с обязательным контрастированием либо МРТ с контрастированием или без такового. МРТ является оптимальным методом визуализации аденомы гипофиза, особенно в сочетании с кистозным компонентом, «пустым» турецким седлом, а также в случае микроаденомы, позволяет точно определить степень вовлечения в процесс латероселлярных структур.
Основным в лабораторной диагностике является определение оценки секреции СТГ. Как правило, у больных акромегалией уровень СТГ натощак повышен, однако в 30–53 % случаев это повышение незначительно, при этом почти у 17 % больных уровень СТГ находится в пределах нормы. Оптимальным является частое (каждые 10–20 мин в течение 24 ч) определение уровня СТГ в крови, что позволяет четко дифференцировать здоровых лиц от больных акромегалией (в норме в 75 % проб содержание СТГ определяется на уровне нижней границы чувствительности метода, в 25 % проб допускаются высокие значения уровня СТГ: секреторные пики в полночь, ранние утренние часы). Однако нередко это практически трудновыполнимо.
В настоящее время надежными альтернативными методами диагностики являются:
— определение СТГ каждые 30 мин в течение 2 ч в сыворотке крови на фоне ОГТТ;
— однократное определение в сыворотке крови уровня ИФР-1 (соматомедина С).
Помимо этого, все пациенты подвергаются осмотру окулиста с целью оценки состояния глазного дна, области перекреста зрительных нервов, проведения периметрии на цвета и белый цвет.
Диагноз акромегалии подтверждается, если:
— базальный уровень СТГ превышает 0,4 нг/мл;
— минимальный уровень СТГ на фоне ОПТ более 1 нг/мл;
— повышен уровень ИФР-1 по отношению к данному полу и возрасту.
Дополнительные методы исследования включают:
— определение в крови уровня пролактина, ТТГ, свТ4, свТ3, АКТГ, свободного кортизола в суточной моче, ЛГ, ФСГ, эстрадиола, тестостерона;
— УЗИ щитовидной железы и органов малого таза;
— рентгенографию органов грудной клетки;
— ЭКГ, ЭхоКГ;
— исследование гликемического профиля, биохимический анализ крови.
В случае подозрения на эктопированную продукцию СТГ (особенно при отсутствии МРТ-признаков аденомы гипофиза) дополнительно необходимо проведение МРТ либо КТ грудной и брюшной полости. В лабораторной диагностике проводится определение содержания СТГ в крови, оттекающей из нижних кавернозных синусов.
Лечение
Как показывает богатый мировой опыт ведения больных акромегалией, все пациенты с верифицированным диагнозом, даже при мягком проявлении заболевания, за исключением очень редких случаев, когда имеются сопутствующие заболевания, вызывающие тяжелое состояние больного с неблагоприятным жизненным прогнозом, должны подвергаться лечению, и весьма активно.
Главные цели лечения акромегалии — устранение клинических симптомов заболевания, нормализация секреции СТГ и ИРФ-1, ликвидация источника избыточной продукции СТГ.
В настоящее время по-прежнему основными методами лечения являются хирургический (в большинстве случаев транссфеноидальная и очень редко — транскраниальная аденомэктомия), медикаментозный и лучевой.
Методы лечения акромегалии:
1. Хирургический (транскраниальная и транссфеноидальная аденомэктомия).
2. Лучевой (дистанционная гамма-терапия, протонотерапия, гамма-нож).
3. Медикаментозный (аналоги соматостатина, агонисты дофамина, антагонисты рецепторов гормона роста).
Основными факторами, определяющими метод лечения, являются: состояние зрения, размер и характер роста аденомы, уровень гормона роста, возраст больного, наличие сопутствующей патологии. Иногда врач вынужден избирать тот или иной вид лечения, учитывая желание больного.
Основные факторы, определяющие выбор метода лечения:
1. Состояние зрения.
2. Размеры и характер роста аденомы.
3. Уровень гормона роста.
4. Возраст больного.
5. Наличие тяжелых сопутствующих соматических нарушений.
6. Желание больного.
Эффективность лечения оценивается по определенным строгим критериям, установленным в мае 2000 года на основании консенсуса 68 ведущих нейроэндокринологов и нейрохирургов мира (табл. 2).
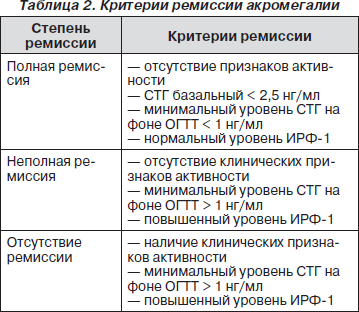
В настоящее время для оценки эффективности длительной терапии аналогами соматостатина дополнительно введено понятие «безопасного» уровня СТГ. Величина данного показателя получена в результате эпидемиологического мультицентрового ретроспективного исследования, проведенного на когорте из 1362 пациентов с акромегалией. В данном исследовании было показано, что если уровень СТГ после терапии был менее 2,5 нг/мл, общий уровень смертности не отличался от такового в общей популяции.
По-прежнему аденомэктомия является первичным методом лечения акромегалии, особенно в случаях, когда имеются признаки хиазмального синдрома, указывающего на сдавление опухолью гипофиза зрительных нервов. Степень восстановления зрения зависит от длительности предшествующей компрессии зрительных нервов и степени выраженности атрофии дисков.
Способ оперативного вмешательства определяется размерами аденомы и особенностями ее распространения.
Главным преимуществом оперативного вмешательства является быстрота наступления эффекта. В случае успешной (радикальной) операции уже в раннем послеоперационном периоде отмечается нормализация ответа СТГ на введение глюкозы (снижение менее 1 нг/мл), а также нормализация уровня ИРФ-1 через несколько недель.
Результаты оперативного вмешательства значительно варьируют и определяются многими факторами. В частности, одним из главных является размер и характер распространения аденомы. Так, наличие у пациента опухоли эндоселлярной локализации позволяет достичь ремиссии в 78–88 % случаев, при этом в случае экстраселлярных аденом, особенно гигантских, этот шанс практически сводится к нулю (табл. 3).
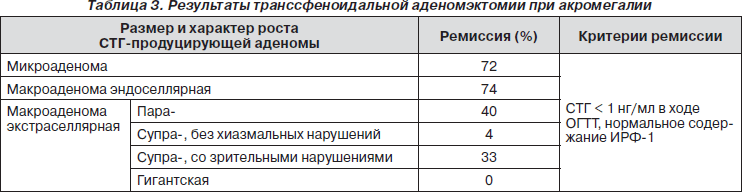
Отсутствие эффекта от оперативного вмешательства может быть также обусловлено инвазией в твердую мозговую оболочку СТГ-продуцирующих аденоматозных клеток, частота которой достигает 43 %, с возникновением истинного рецидива. Повторная операция нужна лишь в том случае, если она будет реальным шансом полного удаления аденомы.
Значительно влияет на исход оперативного вмешательства степень технического нейрохирургического оснащения, в частности использование эндоскопического контроля, нейронавигации; интраоперционный гормональный анализ и МРТ-контроль, а также опыт, квалификация нейрохирурга.
Условия успешной операции:
1. Размеры аденомы:
— микроаденома;
— макроаденома без экстраселлярного распространения.
2. Квалификация нейрохирурга — опыт не менее 200 операций с применением эндоскопического контроля, нейронавигации.
3. Отсутствие морфологических признаков «агрессии» опухоли (атипия клеток, множество митозов).
4. Отсутствие признаков инвазии (инфильтрация или перфорация диафрагмы турецкого седла, твердой мозговой оболочки, кливуса, кавернозных синусов).
В случае проведения операции квалифицированным нейрохирургом послеоперационные осложнения возникают менее чем в 2 % случаев. К ним относятся: зрительные нарушения, менингит, смертельный исход. Такие осложнения, как назальная ликворея, несахарный диабет, гипопитуитаризм, имеют место не более чем у 5 % больных. Если оперативное вмешательство произведено хирургом недостаточной квалификации, процент осложнений возрастает в 3–4 раза. С учетом вышеизложенного, все больные в послеоперационном периоде нуждаются в дополнительном обследовании с целью исключения гипопитуитаризма и при необходимости в адекватной и своевременной его коррекции, а также контрольном осмотре невропатологом, окулистом и отоларингологом, что является важным условием дальнейшей успешной реабилитации.
Уже почти 100 лет лучевая терапия используется в качестве метода лечения заболеваний гипофиза. Впервые А. Beclere в Париже, а Gramagna в Венеции в 1909 году применили данный вид лечения при акромегалии. Самые распространенные виды лучевой терапии — это гамма-терапия и протонотерапия. Традиционная дистанционная гамма-терапия проводится статическим либо ротационным методом. Используются несколько полей облучения: переднее, косое и два латеральных с целью максимальной концентрации дозы в области гипофиза и минимальной в окружающих тканях. Обычная доза облучения — 45–50 Гр в виде 25 фракционных доз. Ежедневная доза — 1,8 Гр 5 дней в неделю в течение 5 недель. Протонотерапия проводится многопольно-конвергентным методом с 15–25 полей в левой височной области способом «напролет» в дозе 50–80 Гр, как правило, в один сеанс.
Ранее широко используемые, данные виды лучевой терапии ввиду их существенных недостатков, с одной стороны, и совершенствования хирургической техники и видов медикаментозной терапии — с другой в настоящее время занимают третье место по значимости в лечении акромегалии. Главные их недостатки — отдаленность во времени производимого ими эффекта и осложнения.
Так, в случае гамма-терапии ингибирующий эффект в отношении секреции СТГ и ИРФ-1 возникает не ранее чем через 2 года от момента проведения облучения. В частности, «безопасный» уровень СТГ (менее 2,5 нг/мл) может быть получен у 36 % больных через 2 года от момента проведения облучения, у 44 % — через 5 лет и у 59 % пациентов — через 10 лет. В отношении нормализации уровня ИРФ-1 были получены следующие результаты: через 3 года — у 27 % больных, через 7 лет — у 53 % и через 10 лет — у 56 % лиц. По другим данным, полученным при анализе результатов гамма-терапии у 560 пациентов со сроками наблюдения от 5 до 15 лет, нормализация уровня ИРФ-1 была достигнута лишь у 199 (36 %) человек. При этом в исследованиях с длительностью периода наблюдения менее 7 лет эффективность составила 29 % (61 человек из 210), с длительностью катамнеза более 10 лет данный показатель возрастает до 39 % (138 человек из 350). Однако с увеличением времени от момента проведения лучевой терапии уменьшается количество пациентов, подвергшихся мониторингу, и количество больных с длительностью наблюдения 15–20 лет составляет не более 10 % от общего количества, ограничивая тем самым возможности для анализа. Более того, пациенты без ремиссии заболевания умирают преждевременно, при этом для длительного наблюдения сохраняются лишь хорошо контролируемые по гормональным критериям пациенты, что, в свою очередь, создает искаженное, ложное оптимистическое мнение об эффективности лучевой терапии. В случае протонотерапии начало супрессивного эффекта отмечается через 6–8 месяцев от момента облучения, в сроки от 3 до 8 лет у 46 % пациентов, значительное снижение уровня СТГ с полной его нормализацией у 39 % лиц.
Гипопитуитаризм — наиболее частое позднее осложнение лучевой терапии. Предполагается, что это является следствием поражения гипоталамуса. Все исследователи указывают на появление вновь обнаруженных случаев гипопитуитаризма у значительного количества больных. Частота данного осложнения зависит от величины фракционной дозы (более 2 Гр), а также предшествующего хирургического вмешательства. Степень гипопитуитаризма положительно коррелирует с длительностью постлучевого периода: через 10 лет у 50–60 % пациентов возникают новые проявления гипопитуитаризма, требующие применения заместительной гормональной терапии. В исследовании, проведенном в условиях ЭНЦ РАМН (Россия, г. Москва), частота вновь выявленного гипопитуитаризма после гамма-терапии составляла: гипотиреоз — 15 %, надпочечниковая недостаточность — 11 %, гипогонадизм — 11 %.
Помимо вышеуказанных осложнений с меньшей частотой возникает поражение сосудов головного мозга, что в 2–4 раза увеличивает риск инсульта и значительно повышает показатель смертности по сравнению как с общей популяцией, так и с пациентами, не получившими данный вид лечения. Причем не выявлено зависимости данного показателя от уровней СТГ и ИРФ-1, размера опухоли, ее распространения за пределы турецкого седла или наличия гипопитуитаризма. В 1–2 % случаев возможно развитие индуцированных злокачественных опухолей мозга, а также височной эпилепсии, поражения зрительных нервов, появление синдрома «пустого» турецкого седла. Есть предположения, что лучевая терапия может приводить к нейропсихологическим изменениям, нарушению когнитивных функций ЦНС: снижению памяти, депрессии и т.д. Однако отсутствует достаточное количество исследований с применением специальных психометрических тестов у идеально контролируемых пациентов, находящихся на заместительной гормональной терапии, включая гормон роста.
В связи с вышеизложенным показания к проведению данных видов лечения весьма ограничены. Гамма-терапия как первичный метод лечения применяется только при невозможности проведения аденомэктомии ввиду отсутствия специализированной нейрохирургической службы либо категорическом отказе больного. Как дополнительный метод — при агрессивных опухолях гипофиза с инвазией в окружающие структуры, включая кавернозные синусы и даже височные доли, в случае неполного удаления аденомы, особенно в сочетании с неблагоприятной гистологической картиной с целью подавления дальнейшей клеточной пролиферации и гиперпродукции СТГ. Протонотерапия применяется как первичный метод лечения при аденоме гипофиза до 1,5 см в диаметре у лиц до 55 лет с относительно невысокой активностью (уровень СТГ не более 20 нг/мл).
Лучевая терапия показана также пациентам, резистентным к терапии аналогами соматостатина либо когда есть серьезные противопоказания к проведению данных видов лечения.
Противопоказаниями для проведения лучевой терапии являются: 1) близкое расположение аденомы к перекресту зрительных нервов, особенно при наличии дефектов полей зрения, т.к. после проведения лучевой терапии возникает отек, способный усугублять имеющиеся нарушения зрения. При наличии такого расположения аденомы перед планируемым облучением идеальным является проведение хирургического лечения (аденомэктомии) с целью удаления супраселлярного компонента опухоли; 2) наличие «пустого» турецкого седла.
Ввиду отсроченного эффекта от лучевой терапии после облучения все больные нуждаются в назначении медикаментозной терапии на длительный период.
В последние годы нашел применение новый метод лучевой терапии — стереотактическая радиохирургия (техника линейного ускорения и гамма-нож). Ее главное отличие от дистанционной гамма-терапии — возможность направить однократно очень большую дозу узким фокусирующим пучком на четко ограниченную зону (участок), что значительно уменьшает число осложнений и повышает эффективность данного вида лучевой терапии. Так, по данным ряда исследователей, ремиссия заболевания может быть достигнута у 54 % лиц через 6 лет после облучения. При этом частота развития гипопитуитаризма не превышает 7 %. Однако, как показал анализ отдаленных результатов данного вида облучения у 270 больных в сроки наблюдения от 6 месяцев до 10 лет, нормализация уровня ИРФ-1 была достигнута лишь у 90 (33 %) пациентов. Сравнительный анализ сходных когорт по длительности катамнеза (от 2 до 10 лет) указывает на практическую идентичность результата: 33 % (77 из 232 человек) в группе больных, получивших в качестве метода лечения стереотактическую радиохирургию, и 29 % (99 из 341) в группе, получившей гамма-терапию. Требуется дальнейшее накопление опыта для разработки четких показаний к проведению данного вида лучевой терапии.
Особенностью состояния после проведения лучевой терапии является развитие постлучевой гиперпролактинемии, частота которой в случае гамма-терапии в два раза выше, чем после проведения протонотерапии (соответственно у 52 и 23 % больных). Назначение небольших доз агонистов дофамина приводит к нормализации содержания пролактина.
В настоящее время в качестве медикаментозной терапии при акромегалии применяются три класса препаратов: агонисты дофамина, длительно действующие аналоги соматостатина и антагонисты рецепторов гормона роста.
Агонисты дофамина применяются в лечении больных акромегалией с 1972 года благодаря способности связываться с дофаминовыми рецепторами 2-го типа на уровне гипофиза и вызывать подавление секреции СТГ у части больных акромегалией. Точный механизм действия остается неясным. Как показал 30-летний клинический опыт, самый древний агонист дофамина — бромкриптин — способен вызывать нормализацию уровня СТГ менее чем у 20 % пациентов и менее чем у 10 % больных нормализацию уровня ИРФ-1. Увеличение дозы препарата выше 20 нг/мл не вызывает усиления его действия. Кроме того, лишь у единичных больных бромкриптин вызывает незначительное уменьшение размера опухоли гипофиза. В настоящее время практически не применяется в мировой практике лечения акромегалии, особенно в качестве монотерапии.
Селективный Д2-дофаминовый агонист квинаголид (норпролак) по сравнению с бромкриптином обладает одновременно более выраженным эффектом в отношении супрессии СТГ, а также меньшими побочными эффектами благодаря отсутствию стимуляции Д1-допаминовых рецепторов. Препарат вызывает нормализацию уровня СТГ и ИРФ-1 не более чем у 40 % больных. Дозы препарата — 0,3 мг в день (0,15 мг 2 раза в день), возможно увеличение дозы до 0,6 мг в день.
Созданный в последние годы новый дофаминовый агонист каберголин (достинекс) обладает более длительным периодом действия (до 72 часов) и значительно меньшим количеством побочных эффектов, чем бромкриптин и квинаголид. В дозе от 1 до 3,5 мг (в среднем — 1,75 мг) в неделю либо 0,5 мг ежедневно терапия каберголином вызывает снижение уровня ИРФ-1 у 47–67 % больных, а его нормализацию — в 28–50 % случаев. Максимальный эффект отмечен в случае смешанных аденом гипофиза (СТГ и пролактин-секретирующих). Ингибирующий эффект каберголина зависит также от степени функциональной активности аденомы, в частности исходного уровня ИРФ-1. Исследования показали, что оптимальный эффект отмечен при исходном уровне ИРФ-1 не более 750 нг/мл. В отличие от бромкриптина терапия каберголином вызывает различной степени уменьшение размера аденомы гипофиза в 16–20 % случаев, а при наличии смешанной аденомы — до 50 % исходного объема опухоли.
Таким образом, показанием к назначению терапии агонистами дофамина является наличие у больного смешанной (СТГ-пролактин-секретирующей) аденомы гипофиза с умеренной функциональной активностью. Препарат можно рекомендовать пациентам, не чувствительным к аналогам соматостатина либо отказавшимся от их применения. Возможна комбинация терапии агонистами дофамина с аналогами соматостатина.
Благодаря успехам фармакологии были созданы длительно действующие селективные аналоги природного соматостатина (октреотид, ланреотид, сандостатин-ЛАР).
Октреотид (сандостатин, SMS-201995) — первый аналог соматостатина, используемый в клинической практике с середины 1980-х годов. Обладает высоким сродством к соматостатиновым рецепторам 2-го подтипа, по своей СТГ-ингибирующей активности превышает нативный гормон в 45 раз. В суточной дозе 100 мкг 3 раза в день подкожно вызывает нормализацию уровня СТГ и ИРФ-1 соответственно у 50 и 40 % пациентов, значительно снижая секрецию данного гормона у 85 % больных. Как показало большое количество исследований, терапия октреотидом приводит к быстрому выраженному клиническому эффекту. Улучшение общего состояния наступает уже через несколько дней от начала лечения. У 95 % больных значительно уменьшается степень проявления таких клинических симптомов, как головная боль, отечность, потливость, артралгии, общая слабость.
В последние годы создана пролонгированная форма октреотида — сандостатин-ЛАР, а также ланреотид (соматулин). Заключение лекарства в специальные микросферы из поли-DL-лактид-когликолид-глюкозного полимера обусловливает их особенности фармакокинетики, благодаря чему количество инъекций в отличие от сандостатина составляет не три в день, а всего 2–3 в месяц (30 мг в/м — одна инъекция) в случае соматулина и одна инъекция в 28 дней (10–30 мг в/м) в случае сандостатина-ЛАР.
В химической структуре ланреотида присутствие группы 3-(2-naftyl)-D-A1a вне кольца привело к более высокой избирательности по отношению к соматостатиновым рецепторам по сравнению с нативным соматостатином и медленному ферментному расщеплению. Длительность действия ланреотида PR достигается при помощи того, что ланреотид помещен на поверхности и внутри биодеградирующих микросфер. Непосредственно после инъекции препарат высвобождается с поверхности микросфер, что сопровождается быстрым подъемом концентрации ланреотида в крови через 2 часа после в/м введения и последующим медленным снижением его уровня в течение примерно 48 часов. Затем происходит постепенное высвобождение ланреотида из микросфер по мере их биологического распада, что сопровождается новым подъемом концентрации ланреотида в крови и сохранением его уровня не менее 1 нг/л на 9–14-й день после инъекции.
При введении сандостатина-ЛАР концентрация препарата также быстро нарастает благодаря высвобождению с поверхости микросфер, затем снижается и остается на низком уровне в течение 7 дней с новым подъемом уровня октреотида в течение 7 дней с последующим сохранением высокой концентрации препарата в течение 28–42 дней.
Терапия аналогами соматостатина приводит к обратному развитию нарушений со стороны сердечно-сосудистой системы: значительному регрессу симптомов акромегалической кардиомиопатии, снижению системной артериальной гипертензии, левожелудочковой гипертрофии, улучшает функциональные гемодинамические параметры. Уменьшается частота ночных апноэ.
Лечение аналогами соматостатина также приводит к быстрому улучшению деятельности сердечно-легочной системы во время физических нагрузок, снижению факторов риска сердечно-сосудистых заболеваний и уменьшению толщины intima media сонных артерий. Достижение стабильно нормального уровня ИРФ-1 и супрессии СТГ ниже 1 нг/мл в течение 1 года на фоне терапии сандостатином-ЛАР приводит к практически полному восстановлению функции миокарда левого желудочка.
Сравнительная оценка эффективности различных аналогов соматостатина указывает на преимущества сандостатина-ЛАР (октреотида-ЛАР) в отношении воздействия на размеры опухоли как в качестве первичной, так и дополнительной терапии.
Увеличение длительности терапии сандостати- ном-ЛАР более трех лет приводит к возрастанию числа пациентов с «безопасным» уровнем СТГ до 72 % и нормализации уровня ИРФ-1 у 79 % лиц, что полностью совпадает с результатами радикального хирургического лечения в случае эндоселлярных аденом. Выявлена прямая корреляционная зависимость длительности терапии аналогами соматостатина и степени снижения ИРФ-1. Более того, не исключается, что длительная терапия сандостатином-ЛАР может приводить к изменению биологической активности молекулы СТГ. Феномена тахифилаксии во время лечения отмечено не было.
Эффективность терапии аналогами соматостатина при акромегалии не зависит от пола пациента, предыдущего хирургического или лучевого лечения. В некоторых исследованиях лучший контроль достигался у пациентов с невысокими исходными гормональными показателями и/или у пожилых пациентов. При помощи регрессионного анализа было показано, что уровень СТГ/ИРФ-1 соответственно через 3–6 месяцев терапии является самым точным прогностическим фактором для финального исхода терапии, более точным, чем базальный уровень СТГ/ИРФ-1 до начала терапии, длительность наблюдения или доза препарата.
Аналоги соматостатина в настоящее время являются препаратами первой линии в медикаментозном лечении акромегалии. Применяются в случае не-удачного хирургического вмешательства и лучевой терапии как дополнительная терапия, особенно если пациент нечувствителен к терапии агонистами дофамина либо они абсолютно противопоказаны. В отличие от агонистов дофамина аналоги соматостатина являются эффективными средствами в качестве первичного метода лечения, особенно у пожилых, ослаб-ленных больных. Наконец, благодаря способности вызывать уменьшение размера опухоли гипофиза они являются эффективным средством предоперационной подготовки.
С начала 2000 года в практику лечения акромегалии вошел принципиально новый препарат — пегвисомант (сомаверт), являющийся генно-инженерным аналогом эндогенного гормона роста с 9 мутациями, антагонистом рецепторов гормона роста. Определяющей функциональные особенности молекулы пегвисоманта является замена глицина на аланин в положении 120 в области связывания (сайта) 2, блокирующая связывание с рецептором. Одновременно восемь аминокислотных замен в области связывания (сайта) 1 значительно повышают аффинность молекулы антагониста к рецептору. Конформационные изменения, возникающие благодаря данной перестройке, приводят к нарушению связывания молекулы антагониста с рецептором, что, в свою очередь, вызывает нарушение процесса димеризации рецептора и индукции синтеза и секреции ИРФ-1. Это позволяет предупредить периферические эффекты избытка СТГ на клеточном уровне вне зависимости от присутствия соматостатиновых или дофаминовых рецепторов в опухоли гипофиза.
Терапия пегвисомантом уже через 2 недели от момента введения препарата приводит к значительному регрессу клинических симптомов, снижению уровня ИРФ-1 на 75 % от исходного, через 3 месяца — к нормализации данного показателя у 82 %, а через 12 месяцев — у 97 % пациентов при коррекции дозы препарата в процессе лечения.
Одним из самых важных эффектов пегвисоманта является его способность к коррекции различных метаболических нарушений, которые всегда имеются у больных акромегалией и являются основной причиной их инвалидизации и повышенной смертности. В частности, данный препарат приводит к ликвидации гиперинсулинемии и снижению инсулинорезистентности, являющейся фактором риска развития сердечно-сосудистых заболеваний. Пегвисомант восстанавливает показатели липидного обмена, а также уровни лептина и показатели костного ремоделирования. Применяется в дозе 10–30 мг подкожно ежедневно. Побочные эффекты минимальные. Могут проявляться покраснением в месте инъекции, незначительной тошнотой (до 10 % больных). В течение первого месяца лечения отмечается существенное повышение уровня СТГ в сыворотке крови, коррелирующее с дозой вводимого препарата без существенного последующего возрастания, несмотря на продолжающуюся терапию. Имеются данные о единичных случаях незначительного продолженного роста аденомы на фоне терапии данным препаратом, преимущественно у лиц, не получивших в анамнезе лучевую терапию. Есть указания на 6 случаев значительного повышения уровня трансаминаз (АЛТ и ACT) через несколько недель терапии, при этом у 4 больных наступила спонтанная нормализация на фоне продолжающейся терапии пегвисомантом, у 2 — нормализация после прекращения лечения. На фоне лечения в 7–16 % случаев отмечается появление антител к СТГ в низком титре, а также антител к пегвисоманту, без отрицательного влияния на степень нормализации ИРФ-1. С учетом вышесказанного при назначении терапии пегвисомантом необходим контроль размеров аденомы гипофиза (1 раз в 6 месяцев), функции печени (1 раз в месяц). Контроль уровня ИРФ-1 необходимо проводить каждые 4–6 недель с возможной коррекцией дозы препарата. Начальная доза составляет 10 мг в сутки, максимальная ежедневная доза 30 мг.
Несмотря на высокую эффективность препарата, ввиду недостаточной изученности его безопасности и действия на размеры опухоли гипофиза, показаниями к его применению являются: 1) отсутствие эффективности аналогов соматостатина при сроке лечения ими не менее 3 месяцев; 2) сохранение выраженных побочных эффектов после 6 и более месяцев терапии аналогами соматостатина, несмотря на их клинико-гормональную эффективность.
В 2009 г. опубликован консенсус по лечению акромегалии, в котором представлен ступенчатый алгоритм ведения пациентов с СТГ-секретирующей аденомой гипофиза (рис. 3).
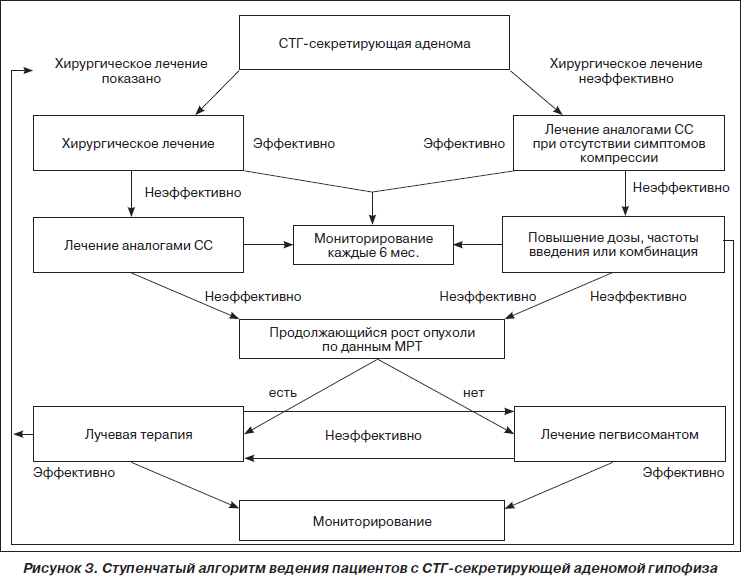
Симптоматическая терапия акромегалии определяется в каждом конкретном случае и зависит от характера и степени вовлечения в патологический процесс того или иного органа или системы организма (коррекция изменений со стороны сердечно-сосудистой системы, опорно-двигательного аппарата, метаболических нарушений, лечение осложнений лучевой терапии).
Оценка эффективности лечения
Для оценки эффективности проводимой терапии контроль уровня СТГ и ИФР-1 проводится через 3–6–12 месяцев от начала терапии, МРТ-контроль — через 6–12 месяцев.
Осложнения и побочные эффекты лечения
Возможные побочные эффекты терапии аналогами соматостатина в порядке убывания распространенности:
— диарея;
— метеоризм;
— боли в животе;
— стеаторея;
— тошнота.
Частота перечисленных явлений составляет 8– 45 %, причем эти эффекты наиболее выражены после первой инъекции, при продолжении лечения через 6 месяцев частота побочных эффектов уменьшается в 3–7 раз.
Кроме того, у 2–9 % пациентов могут появляться бессимптомные камни в желчном пузыре, застой желчи, расширение желчного протока.
Тем не менее многочисленные исследования доказали безопасность лекарственных средств данной группы в качестве средств для длительной терапии.
Ошибки и необоснованные назначения
Несмотря на высокую эффективность пегвисоманта, в связи с недостаточной изученностью его безопасности и действия на размеры опухоли гипофиза показания к его применению пока ограничены.
Агонисты дофаминовых рецепторов абсолютно противопоказаны при язвенной болезни желудка и двенадцатиперстной кишки.
Прогноз
Прогноз зависит от своевременности выявления и успешности лечения заболевания. При применении октреотида ремиссия заболевания возможна в 70–80 % случаев, октреотида длительного действия — в 88 %, ланреотида — в 70 %. При использовании пегвисоманта ремиссии удается добиться у 97 % больных. Неселективные дофаминомиметики (при приминении в качестве монотерапии) приводят к ремиссии лишь в 10–20 % случаев, хинаголид — в 40 %. При применении каберголина ремиссия заболевания возможна до 53 % случаев (при смешанной, СТГ-пролактинсекретирующей аденоме — до 67 %). Возможно уменьшение размера аденомы гипофиза в 16–20 % случаев, а при наличии смешанной аденомы — до 50 % исходного объема опухоли.
Профилактики данного заболевания не существует.


1. Караченцев Ю.И., Хижняк О.О., Микитюк М.Р., Куцын В.Н. Акромегалия и гигантизм. — Киев: Старт-98, 2010. — 132 с.
2. Дедов И.И., Молитвословова Н.Н., Марова Е.И. Акромегалия: патогенез, клиника, диагностика, дифференциальная диагностика, методы лечения: Пособие для врачей. — Тверь: ООО «Издательство «Триада», 2006. — 48 с.
3. Эндокринология / Под ред. проф. П.Н. Боднара. — Винница: Нова книга, 2007. — 344 с.
4. Сергієнко О.О. Основи захворювань гіпоталамо-гіпофізарної системи. — Львів: Атлас, 2002. — 116 с.
5. Рациональная фармакотерапия заболеваний эндокринной системы и нарушений обмена веществ / Под общ. ред. И.И. Дедова, Г.А. Мельниченко. — Москва: Литтерра, 2006. — С. 429-439.
6. Мельниченко Г.А., Марова Е.И., Дзеранова Л.К. Диагностика и лечение нейроэндокринных заболеваний. — М.: Адамантъ, 2002.
7. Guidelines for acromegaly management / S. Melmed, A. Colao, A. Barkan et al. // J. Clin. Emdocrinol. Metab. — 2009. — Vol. 94, № 5. — P. 1509-1517